Changes triggered by COVID19 in your quality system
The 2020 global pandemic has changed life as we know it, but this article focuses on three crucial quality system changes triggered by COVID19.
Last night my daughter Gracie mentioned that her teacher assigned an essay to write about three changes triggered by COVID19 in her life. The three things that she felt had changed the most were: 1) she goes to bed much later, and sleeps in every day; 2) her school is closed, and she only talks to her teacher twice per week via Zoom, and 3) she misses her friends. I know that her story is similar to my son Bailey who is in his Freshman year of college, and I know that my personal story is quite similar. Coincidentally, I started writing this article earlier this week about three significant-quality system changes triggered by COVID19:
- If you are going to conduct on-site audits, you need to ask about using personal protective equipment (PPE).
- There needs to be a greater focus on business continuity plans and robust supply chain monitoring.
- Remote audits are suddenly encouraged for 1st, 2nd, and 3rd-party audits.

Changes triggered by COVID19: #1 Use Face Masks
US FDA Issues EUAs
At the beginning of the COVID19 pandemic, the US FDA created several emergency use authorizations (EUA). The three EUA areas were IVD testing, ventilators, and face masks. The EUA for IVD testing is not surprising, because the FDA issues and EUA every time a new lethal and contagious virus emerges (e.g., Zika and Ebola). The EUA for ventilators was issued because the number of people with respiratory issues was expected to explode with the spread of the virus, and the supply chain for components of ventilators had already been disrupted by the initial spread of the virus in China. The EUA for face masks was issued because it is the second-best way to protect people from the virus, and existing infrastructure for face mask production could not possibly supply the entire world with face masks overnight.
Everyone in the World Gets a Face Mask
As soon as the EUA for face masks was issued, every regulatory consultant in the USA was inundated with urgent requests for help to complete EUA requests for masks. I also received similar requests for assistance with Canadian filings. The FDA did a great job of providing detailed information about the different types of face masks (i.e., face masks, surgical face masks, and N95 respirators). Testing companies created new website pages specifically for each of the different face mask tests, and every company with a sewing machine suddenly wanted to manufacture masks. I even read an article about an elderly woman making face masks for her entire family while she listened to The Beatles “HELP!” in the background.
Why aren’t you wearing your face mask?
Even after the world makes the first 7 billion face masks, not everyone will wear their face masks. Masks will protect us from touching our hands to our face–which spreads many germs in addition to the SARS-CoV-2 virus. Masks will also keep us from coughing on other objects and people if we have the virus. Finally, face masks protect us from the small droplets that carry the virus from one person to the next. Even though there are obvious safety reasons for everyone in the world to wear a face mask, most people don’t want to wear a face mask. This is no different from the argument to wear a seat belt, and unless our government creates a law or temporary order requiring us to wear face masks, most people won’t bother to wear one.
Changes triggered by COVID19: Auditors need to wear face masks
As a medical device auditor, I feel I must always follow the safety rules in every facility I visit. Lead auditors are supposed to contact the company ahead of time and ask about the safety policies as part of audit preparation and initiating the audit. I’m 6’6” (2.00m) in height, and my shoe size is 14. There is seldom gowning for me to wear that fits appropriately–especially in Southeast Asia. I squeeze into the garments, and they are uncomfortable and hot, but I wear the garments anyway. My job includes auditing clean rooms, and I can’t do my job without gowning up. By following the rules, I also eliminate the excuses for anyone in the facility I visit. Now that we have a global pandemic, you should be wearing a face mask in every medical device facility to protect yourself, people you work with, and users of medical devices. You should also consider carrying spare face masks with you to protect yourself on airplanes, in hotels, etc.
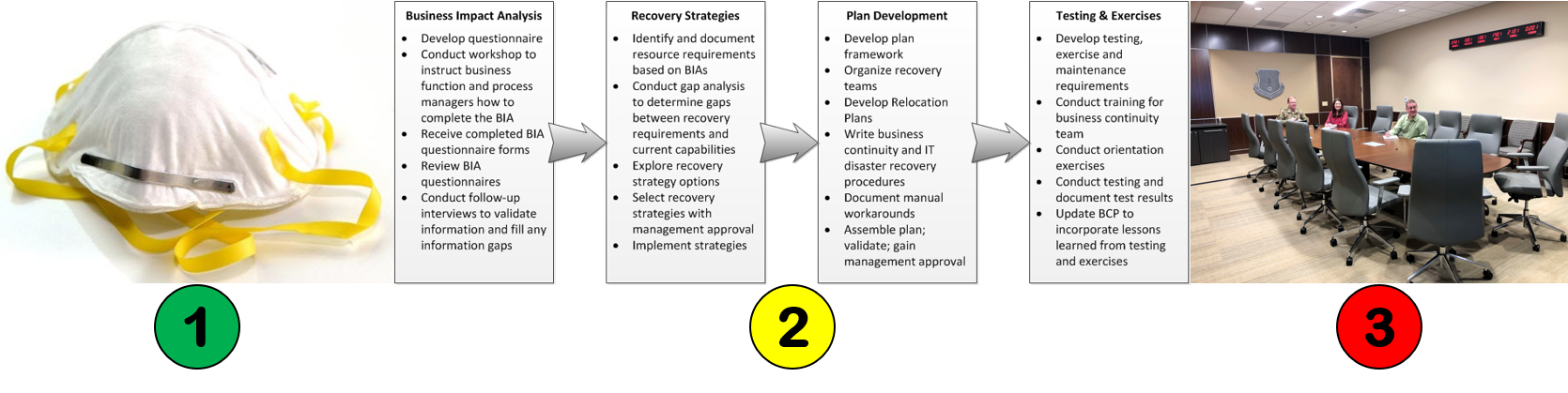
Changes triggered by COVID19: #2 Business Continuity Plans
Will business continuity plans be required now?
In addition to the cultural shift to wearing face masks, we will also need to make significant changes in our overall preparations for natural disasters, fires, and biological threats. Although there is no specific requirement for a business continuity plan in ISO 13485:2016, there are many places where an auditor can identify a requirement to maintain the effectiveness of a quality system (no exceptions):
- Clause 1, Scope
- Clause 4.1.1 & 4.1.3, General Quality System Requirements
- Clause 5.3, Quality Policy
- Clause 5.4.2, Quality management system planning
- Clause 5.6.3, Management Review Output
- Clause 6.1, Provision of resources
- Clause 8.1, General requirements for Measurement, analysis, and improvement
- Clause 8.2.4, Internal audit
- Clause 8.5.1, General Improvement
Although any of these clauses could potentially be referenced as a requirement for a business continuity plan, the last clause would generally be the most appropriate. This clause states, “The organization shall identify and implement any changes necessary to ensure and maintain the continued suitability, adequacy and effectiveness of the quality management system…”. In this time of radical change, adding provisions to your business continuity plan for coping with a global biological threat seems obvious and urgently needed.
Suggested content for your business continuity plan
Sadly, the USA was probably better prepared for a disaster in the 1960s after the Cuban Missile Crisis than we are today. If you do not yet have a business continuity plan, or if you need suggestions for improving your plan, the following is a list of suggested items to include in your plan:
- Develop a plan for power outages, fires, floods, earthquakes, severe wind/tornadoes, hurricanes, workplace violence, and biological threats
- Develop an emergency alert system to notify employees of any emergency
- Build emergency kits and store the kits for when they are needed
- Document your plan in multiple formats (virtual and physical) and distribute to all employees–including a social media plan
- Translate your plan into multiple languages for non-English speaking employees
- Develop a training program that addresses the various aspect of emergency preparation
- Practice your plan just like fire drills, so everyone is prepared and nobody panics
The Ready.gov website has many resources for the above items, including a series of “Ready Business Videos” and “Ready Business Toolkits.”
How to practice your business continuity plans
My sister is a teacher, and she is in the process of opening a new charter school in Maine. We were discussing her planning for the school, and the disruption of schools by the COVID19 pandemic has challenged all teachers to learn to use distance learning. My sister’s school focuses on teaching children about the environment, and she doesn’t like to spend lots of time on the computer. I was sharing some of the environmental studies my daughters are receiving via Zoom from their teachers. I suggested that she might want to pick one topic each week to teach via distance learning. The purpose of this would be to give her, and her students practice using distance learning for a variety of subjects. Therefore, when we experience another biological disaster, her students will already know precisely how to use distance learning to continue their education. My argument was that this routine use of distance learning would be a more effective preparation for emergencies than a once-per-month “fire drill.” Companies should use the same approach. Your company should create a schedule for practicing remote management meetings and working from home. This will ensure that systems are in place to keep your business running smoothly when disaster strikes again.
Changes triggered by COVID19: Expect regulators to require business continuity plans
The widespread shortage of face masks, ventilators, and other critical supplies needed during the COVID19 pandemic is going to result in new regulations requiring business continuity plans. This is a certainty born from the observation that every single medical device regulation we have resulted from severe public health threats. The COVID19 pandemic is the biggest global health crisis the world has experienced in 100 years. Therefore, we can expect corrective actions in the form of new regulations requiring companies to have a business continuity plan. Some regulators will act independently, but I would expect this also to be an action taken by the International Medical Device Regulators Forum (IMDRF). We can also expect there to be new laws requiring amendments to business continuity plans for public companies. The Sarbanes-Oxley Act of 2002 requires public companies in the USA to have business continuity plans. Despite this requirement, many public companies have been ruined by the COVID19 pandemic. Therefore, we should expect amendments to these requirements and revisions to the international standard for business continuity planning (i.e., ISO 22301:2019). We should also expect to see new interest in becoming certified to this standard.
Changes triggered by COVID19: #3 Remote Auditing
What are certification bodies doing about surveillance audits and re-certification audits?
Most of the companies that had initial certification audits scheduled for the first quarter of 2020 were forced to reschedule their audits because the employees must work from home, and the certification bodies must conduct at least some of their audits on-site. The FDA was also forced to cancel all foreign inspections temporarily. However, companies that already have certification need surveillance audits and re-certification audits to maintain the validity of their quality system certificates. Therefore, certification bodies now have plans for conducting audits remotely. For companies that virtual medical device manufacturers, certification bodies can conduct full quality system audits remotely. However, manufacturers with production activities on-site are only able to conduct partial audits. The certification bodies must still conduct on-site audits, but they are being permitted six months to conduct an on-site audit to cover the gaps remaining from the partial remote audits. Prior to conducting the partial remote audits, certification bodies are sending out questionnaires to all of their clients to gather information about whether the manufacturers can support a remote audit and to what degree.
Second-party audits conducted remotely
Second-party audits, also known as supplier audits, have always been of interest for manufacturers to conduct remotely–mainly if the supplier is located overseas. The US FDA regulations do not require companies to conduct supplier audits. However, if there are quality problems with suppliers, you are expected to conduct a thorough investigation to identify the root cause of quality problems, in most cases, that require an on-site audit. However, if your suppliers are providing good quality and they are ISO 13485:2016 certified, then you probably are using this as a justification for not conducting on-site audits or at least reducing the frequency of those audits. Now that most people are not able to travel, or because the people you need to speak with are working from home, manufacturers are being forced to conduct remote audits. This has always been permitted, but the effectiveness of remote audits is often questioned. Supply chain disruptions are now a global issue that is impacting the safety and effectiveness of our hospitals, and regulators will expect you to improve the rigor of your supplier evaluations–including conducting more supplier audits. Therefore, establishing more effective procedures for remote supplier auditing is urgently needed.
Changes triggered by COVID19: We need to develop procedures for remote auditing
Although most first-party audits are conducted on-site, especially if conducted by employees of your company, we will still need to establish procedures for remote auditing for internal audits. Some of our client’s scheduled internal audits for April and May that they had to cancel because they were unable to access the records needed for the audit while they were working from home. In addition, most of the US States have implemented stay-at-home audits that prevent our team from traveling to our clients. This is forcing our team to develop more robust procedures for remote auditing. We needed to change our audit agendas to accommodate eight 90-minute audit sessions in four days, instead of conducting two full days of on-site auditing. We are also doing more preparation before the audit to allow the auditees time to scan paper records so that we can review those records remotely. Finally, we are experimenting with techniques for collaboration as an audit team so that multiple auditors can simultaneously audit a client and complete a full quality system audit more quickly without forcing any one person to work for longer than 90 minutes in front of a computer. We are still perfecting these new methods, but we are writing a series of articles on this topic. You can order the book from Amazon ($5 pre-order discount until August 28, 2020).
Thank you & Future Articles
Thank you for reading. This is the longest article we have published on our site since 2012. This article also kicks off a ten-part blog series specific to remote auditing techniques:
- Remote audit opening meeting – 4 changes – May 12
- Audit team communications – May 19
- Remote audit resources – software and hardware tools – May 26
- How to apply a risk-based auditing approach to audits and remote audits – June 2
- How to make a supplier questionnaire for remote auditing – June 25
- Remote audit duration less than 90 minutes – June 30
- Remote auditing work instruction – July 14
- Planning partial remote audits – July 21
- Remote audit invitations – 4 things to remember – August 4
- Training new audit team members and lead auditors – August 11
There are also five new live webinars planned on related topics:
- Opening Meetings Webinar (free) – May 14, 2020
- Audit team communication during a remote audit (free) – June 4, 2020
- How to qualify your supplier’s Webinar (pre-order by June 1) – June 25, 2020
- Remote auditing techniques webinar (pre-order by July 1) – July 16, 2020
- MDSAP Certification Body Interviews (free) – August 6, 2020
Changes triggered by COVID19 in your quality system Read More »