Learn how to become ISO 13485 certified while avoiding the stress that tortures other quality system managers.
What is ISO 13485?
ISO 13485 is an international standard for quality management systems that is specific to the medical device industry. ISO 13485:2016 is the most recent version of the standard, and it has become the blueprint for medical device company quality systems globally. If your company wants to design, manufacture, or distribute medical devices, you should consider becoming ISO 13485 certified.
Do you have to purchase a copy of ISO 13485?
Yes, you need to maintain a copy of the ISO 13485 standard as a “document of external origin.” This is needed for reference when you are making updates to procedures in your quality system. If you are looking for the best place to purchase a copy of the ISO 13485:2016 standard, we recommend the Estonian Centre for Standardisation and Accreditation. If you purchase a copy, we recommend selecting the option for a multi-user license so the standard can be used by more than one person in your company and printed. The only difference between the EN ISO version and the International ISO version is that the EN ISO version includes harmonization Annex ZA for compliance with the EU MDR and Annex ZB for compliance with the EU IVDR. This version is also referred to as A11:2021. Here’s a copy of the text from the beginning of the Standard:
“This Estonian standard EVS-EN ISO 13485:2016/A11:2021 consists of the English text of the European standard EN ISO 13485:2016/A11:2021. This standard has been endorsed with a notification published in the official bulletin of the Estonian Centre for Standardisation and Accreditation. Date of Availability of the European standard is 08.09.2021. The standard is available from the Estonian Centre for Standardisation and Accreditation.”

Medical Device Academy’s experience with ISO 13485 training
Rob Packard created his first quality system in the Spring of 2004. In October 2009, after successfully managing quality systems for three different medical device manufacturers, Rob joined BSI as a Lead Auditor and instructor. In April 2010, he purchased the 13485cert.com URL and he began to help companies implement quality systems as a consultant (while continuing to audit and train 140 days per year for BSI). In 2011 his medical device blog postings began as a way to help medical device companies. In 2012, Rob began building a library of quality system procedures for a turn-key quality system and selling the procedures from the Medical Device Academy website. Dozens and dozens of consulting clients have successfully achieved ISO 13485 certification with Medical Device Academy’s turnkey quality system procedures, and hundreds of quality systems were audited and/or improved. This ISO 13485 training webinar is also included as part of our turnkey quality system.
Projected Changes for the FDA’s update to the QMSR
On February 23, 2022, the FDA published a proposed rule for medical device quality system regulation amendments. The FDA planned to implement amended regulations within 12 months, but the consensus of the device industry is that a transition of several years would be necessary. In the proposed rule, the FDA justifies the need for amended regulations based on the “redundancy of effort to comply with two substantially similar requirements,” creating inefficiencies. The FDA also provided estimates of projected cost savings resulting from the proposed rule. What is completely absent from the proposed rule is any mention of the need for modernization of device regulations.
On January 31, 2024, the FDA issued a final rule amending the device current good manufacturing practice (CGMP) requirements of the Quality System (QS) Regulation under 21 CFR 820 to align more closely with ISO 13485. The FDA will begin enforcing this on February 2nd, 2026.
The QSR is 26 years old, and the regulation does not mention cybersecurity, human factors, or post-market surveillance. Risk is only mentioned once by the regulation, and software is only mentioned seven times. The FDA has “patched” the regulations with guidance documents, but there is a desperate need for new regulations that include critical elements. The FDA has “patched” the regulations through guidance documents, but there is a desperate need for new regulations that include critical elements. The transition of quality system requirements for the USA from 21 CFR 820 to ISO 13485:2016 will force regulators to establish policies for compliance with each of these quality system elements. Companies that do not already have ISO 13485 certification should be proactive by 1) updating their quality system to comply with the standard and 2) adopting the best practices outlined in the following related standards:
- AAMI/TIR57:2016 – Principles For Medical Device Security – Risk Management
- IEC 62366-1:2015 – Medical devices — Part 1: Application of usability engineering to medical devices
- ISO/TR 20416:2020 – Medical devices — Post-market surveillance for manufacturers
- ISO 14971:2019 – Medical Devices – Application Of Risk Management To Medical Devices
- IEC 62304:2015 – Medical Device Software – Software Life Cycle Processes
- ISO/TR 80002-1:2009 – Medical device software — Part 1: Guidance on the application of ISO 14971 to medical device software
- ISO/TR 80002-2:2017 – Medical device software — Part 2: Validation of software for medical device quality systems
Previous versions of the ISO 13485 webinars
This 2-part webinar has been previously recorded three different times. Our previous webinar on the 2003 version of ISO 13485 was split into two parts: Stage 1 and Stage 2. That first webinar was recorded in 2015. The webinars were updated in 2016 and again in 2018. We followed the same format, 2-part Stage 1 and Stage 2, for all of the subsequent ISO 13485 training webinars. Stage 1 is a preparatory review, whereas Stage 2 is the in-depth validation for actual certification. Stage 1 is usually 1 to 1.5 days and can be conducted remotely. Stage 2 is usually 3 or more days in duration.
The Stage 1 certification audit training focuses on the following processes:
- Introduction (0)
- Scope (1)
- Normative References (2)
- Terms and definitions (3)
- QMS General Requirements (4.1)
- Quality System Documentation (4.2)
- Management Commitment (5.1)
- Customer Focus (5.2)
- Quality Policy (5.3)
- Quality Objectives (5.4.1)
- Quality Planning (5.4.2)
- Management Responsibility, Authority, and Communication (5.5)
- Management Review (5.6)
- Provision of Resources (6.1)
- Measurement, Analysis, and Improvement (8.1)
- Feedback (8.2.1)
- Internal Auditing (8.2.4)
- Monitoring & Measurement of Processes (8.2.5)
- Monitoring & Measurement of Product (8.2.6)
- Data Analysis (8.4)
- General Improvement (8.5.1)
- Corrective Action (8.5.2)
- Preventive Action (8.5.3)
The Stage 2 certification audit training focuses on the clauses of the ISO 13485:2016 Standard that were not covered in the Stage 1 readiness audit:
- Medical Device File (4.2.3)
- Document Control, including Documents of External Origin (4.2.4)
- Control of Records (4.2.5)
- Human Resources/Training (6.2)
- Infrastructure (6.3)
- Contamination Control (6.4)
- Planning of Product Realization (7.1)
- Customer-Related Processes (7.2)
- Design Controls (7.3)
- Purchasing (7.4)
- Production Process Controls (7.5.1)
- Cleanliness (7.5.2)
- Installation (7.5.3)
- Servicing (7.5.4)
- Sterile Product Requirements (7.5.5)
- Process Validation (7.5.6)
- Sterilization Validation & Sterile Barrier Validation (7.5.7)
- Identification (7.5.8)
- Traceability (7.5.9)
- Customer Property (7.5.10)
- Preservation of Product (7.5.11)
- Control of Monitoring & Measurement Devices (7.6)
- Complaint Handling (8.2.2)
- Reporting to Regulatory Authorities (8.2.3)
- Control of Nonconforming Product (8.3)
The most recent version of ISO 13485 webinars
This new version of ISO 13485:2016 training will be delivered as a free live-streaming video. After live-streaming the videos, they will remain available to watch as refresher training. The dates of the training sessions are:
- Stage 1 – live-streaming on September 12, 2025 @ Noon ET
- Stage 2 – live-streaming on September 19, 2025 @ Noon ET
This pair of ISO 13485 training webinars explains precisely what you need to do to implement a quality system compliant with ISO 13485. After you creating your own plan (a free template is provided of you register to take the training quiz associated with this ISO 13485:2016 training. You can watch these two webinars with your team so they can implement your plan in the next several months. All deliveries of content are through our streaming channels (e.g., YouTube).
Exam and Training Certificate available
After completing each training webinar, you can verify the training effectiveness and obtain training records from us if you complete the registration form for this webinar above. You can watch the training anytime, watch as many times as you wish, and you can complete the training quizzes anytime. The quizzes will be available after each live-streaming video airs.

There is a big difference between being ISO 13485 certified and being compliant with ISO 13485:2016, the medical devices quality management systems standard. Anyone can claim compliance with the standard. Certification, however, requires that an accredited certification body has followed the requirements of ISO 17021:2015, and they have verified that your quality system is compliant with the standard. To maintain that certification, you must maintain your quality system’s effectiveness and endure both annual surveillance audits and a re-certification audit once every three years.
Step 1 – Planning for ISO 13485 certification
There are six steps in the ISO 13485 certification process, but that does not mean there are only six tasks. The first step in every quality system is planning. Most people refer to the Deming Cycle or Plan-Do-Check-Act (PDCA) Cycle when they describe how to implement a quality system. However, when you are implementing a full quality system, you need to break the “doing” part of the PDCA cycle into many small tasks rather than one big task. You also can’t implement a quality system alone. Quality systems are not the responsibility of the quality manager alone. Implementing a quality system is the responsibility of everyone in top management.
Below you will find seven tasks listed. I did NOT identify these nine tasks as “Steps” in the ISO 13485 certification process, because these tasks are typically repeated for each process in your quality system. Most quality systems are implemented over time, and the scope of the quality system usually grows. Therefore, you are almost certain to have to perform all of the following nine tasks multiple times–even after you receive the initial ISO 13485 certification. As the saying goes, “How do you eat an elephant? One bite at a time.” Therefore, avoid the inevitable heartburn caused by trying to do too much at one time. Implement your quality system one “bite” at a time.
Task 1 – Purchase applicable standards
The first task in implementing an ISO 13485 quality system is to purchase a copy of the ISO 13485:2016 standard, such as the MDSAP Audit Approach document. You will also need other applicable medical device standards. Some of these standards are general standards that apply to most, if not all, medical devices, such as ISO 14971:2019 for risk management. There are also guidance documents that explain how to use these general standards, such as ISO/TR 24971:2020, and guidance on how to apply ISO 14971:2019. Finally, there are testing standards that identify testing methods and acceptance criteria for things such as biocompatibility and electrical safety. You will need to monitor these standards for new and revised versions. When these standards are updated, you will need to identify the revised standard and develop a plan for addressing the changes.
When you purchase a standard, be sure to buy an electronic version of the standard so you can search the standard for keywords efficiently. You should also consider purchasing a multi-user license for the standard because every manager in your company will need to look up information in the standard. Alternatively, you could buy a paper copy of the standard and locate the standard where everyone in your company can access it. Often I am asked what the difference is between the EN version of the standard and the ISO version of the standard. “EN” is an abbreviation meaning European Standards or “European Norms,” which is based upon the literal translation from the French (i.e., “normes”) and German (i.e. “norm”) languages. “ISO” versions are international standards. In general, the body of the standard is typically identical but harmonized EN standards for medical devices include annexes ZA, ZB, and ZC that identify any deviations from the requirements in three medical device directives (i.e., MDD, AIMD, and IVDD).
Task 2 – Identify which processes are applicable
Clause 1 of ISO 13485 is specific to the scope of a quality system. ISO 9001, the general quality system standard, allows you to “exclude” any clause from your quality system certification. However, ISO 13485 will only allow you to exclude design controls (i.e., clause 7.3). Other clauses within ISO 13485 may be identified as “non-applicable” based on the nature of your medical device or service. You must also document the reason for non-applicability in your quality manual. Typically, the following clauses are common clauses identified for non-applicability:
- Clause 4.1.6 – quality system software
- Clause 6.4 – work environment
- Clause 7.5.2 – cleanliness of the product
- Clause 7.5.3 – installation
- Clause 7.5.4 – servicing
- Clause 7.5.5 – sterile devices
- Clause 7.5.6 – process validation
- Clause 7.5.7 – sterilization validation
- Clause 7.5.9.2 – implantable devices
- Clause 7.5.10 – customer property
- Clause 8.3.4 – rework
Task 3 – Assign a process owner to each process
The third task is to assign a process owner to each of the processes in your quality system. Typically, you create a master list of each of the required processes. Usually, the assignments are made to managers in the company who may delegate some or all of a specific process. You should expect most managers to be responsible for more than one process because there are 28 required procedures in ISO 13485:2016, but most companies have fewer than ten people when they first implement a quality system.
Task 4 – Prioritize and schedule the implementation of each process
The fourth task is to identify which processes need to be created first and to schedule the implementation of procedures from first to last. You can and should build flexibility into the schedule, but some procedures are needed at the beginning. For example, you need document control, record control, and training processes to manage all of your other procedures. You also need to implement the following processes to document your Design History File (DHF): 1) design controls, 2) risk management, 3) software development (if applicable), and 4) usability. Therefore, these represent the seven procedures that most companies will implement as early as possible. Procedures such as complaint handling, medical device reporting, and advisory notice procedures are usually reserved for last. These procedures are last because they are not needed until you have a medical device in use.
Task 5 – Create forms, flowcharts, and procedures for each process
Forms create the structure for records in your quality system, and a well-designed form can reduce the need for lengthy explanations in a procedure or work instruction. Therefore, you should consider developing forms first. The form should include all required information that is specified in the applicable standard or regulations, and the cells for that information should be presented in the order that the requirements are listed in the standard. You might even consider numbering the cells of the form to provide an easy cross-reference to the corresponding section of the procedure. Once you create a form, you might consider creating a flowchart next. Flowcharts provide a visual representation of the process. You might consider including numbers in the flow chart that cross-reference to the form as well.
Once you have created a form and a flowchart, you are now ready to write your quality system procedure. Many sections are typically included in a procedure template. It is recommended that you use a template to ensure that none of the basic elements of a procedure are omitted. You might also consider adding two sections that are uncommon to a procedure: 1) a risk analysis of the procedure with the identification of risk controls to prevent risks associated with the procedure, and 2) a section for monitoring and measurement of the process to objectively measure the effectiveness of the process. These metrics are the best sources of preventive actions, and some of the metrics might be potential quality objectives to be identified by top management.
Task 6 – Perform a gap analysis of each procedure
Most companies rely upon internal audits to catch missing elements in their procedures. However, audits are intended to be a sampling rather than a 100% comprehensive assessment. Therefore, when a draft procedure is being reviewed and approved for the first time, or a major rewrite of a procedure is conducted, a thorough gap analysis should be done before the approval of the draft procedure. Matthew Walker created an article explaining how to conduct a gap analysis of procedures. In addition, Matthew has been gradually adding cross-references to ISO 13485:2016 requirements in each procedure. He is color-coding the cross-referenced clauses in blue font as well. This makes it much easier for auditors to verify that a procedure is compliant with the regulations with minimal effort. The success of these two methods has taught us the importance of conducting a gap analysis of all new procedures.
Task 7 – Train applicable personnel for each process
You are required to document the training requirements for each person or each job in your company. Documentation of training requirements may be in a job description or within a procedure. In addition to defining who should be trained, you also need to identify what type of training should be provided. We recommend recording your training to ensure that new future employees receive the same training to ensure consistency. Design controls training should be the first priority. You are also required to maintain records of the training. You must verify that the training was effective, and you need to check whether the person is competent in performing the tasks. This training may require days or weeks to complete. Therefore, you may want to start training people several weeks before your procedure is approved. Alternatively, you can swap the order of tasks and conduct training after the procedure approval. If that approach is taken, then the procedure should indicate the date the procedure becomes effective–typically 30 days after approval to allow time for training.
Task 8 – Approve the procedure
Approval of a procedure may be accomplished by signing and dating the procedure itself, while another approach is to create a document that lists all the procedures and forms being approved at one time. The second method is the method we use in our turnkey quality system. Companies can review and approve as many procedures at one time as they wish. Since this process needs to be defined to ensure that all of the procedures you implement are approved, the document control process is typically the first procedure that companies will approve in a new quality system. The second procedure generally is for the control of records. Then the next procedures implemented will typically be focused on the documentation of design controls, risk management, usability testing, and software development. The last procedures to be approved are typically complaint handling, medical device reporting, and recalls. These procedures are left for last because you don’t need them until you are selling your medical device.
Task 9 – Start using the procedure and generating records
The last task required for the implementation of a new quality system is to start using the procedures to generate records. All of the procedures will need records before the process can be verified to be effective. Records can be paper-based, or the records can be electronic. Whichever format you use for the record retention needs to be communicated to everyone in the company through your Control of Records procedure and/or within each procedure. If you include the information in each procedure, the records of each procedure should be listed in the procedure, and the location where those records are stored should be identified. Generally, there is no specific minimum number of records to have for a certification audit, but you should have at least a few records for each process that you implement.
Step 2 – Conducting your first internal audit
The purpose of the internal audit is to verify the effectiveness of the quality system and to identify nonconformities before the certification body auditor finds them. To successfully achieve this secondary objective, it is essential to have a more rigorous internal audit than you expect for the certification audit. Therefore, the internal audit should be of equal duration or longer in duration than the certification audit. The internal audit should not consist of a desktop review of procedures. Reviewing procedures should be part of gap analysis (i.e., task 6 above) that is conducted on draft procedures before they are approved. Internal audits should utilize the process approach to auditing, and the auditor should apply a risk-based approach (i.e., focus on those processes that are most likely to contribute to the nonconforming products, result in a complaint, or cause severe injuries and death).
After your internal audit, you will receive an internal audit report from the auditor. You should also expect findings from the internal auditor, and you should expect opportunities for improvement (OFI) to be identified. Experienced auditors can typically identify the root cause of a nonconformity more quickly than most process owners. Therefore, it is recommended for each process owner and subject matter expert to review nonconformities with the auditor and discuss how the nonconformity should be investigated. The root cause must be correctly identified during the CAPA process, and the effectiveness check must be objective to ensure that problems do not recur.
Step 3 – Initiating corrective actions
Corrective actions should be initiated for each internal audit finding immediately, to make sure the findings are corrected and prevented from repeat occurrence before the Stage 1 audit. It will take a minimum of 30 days to implement the most corrective actions. Depending upon the scheduling of the internal audit, there may not be sufficient time to complete the corrective actions. However, you should at least initiate a CAPA for each finding, perform an investigation of the root cause, and begin to implement corrective actions.
Also, to take corrective actions related to internal audit findings, you should look for internal audits from other sources. The diagram below shows several different sources of potential corrective and preventive actions.
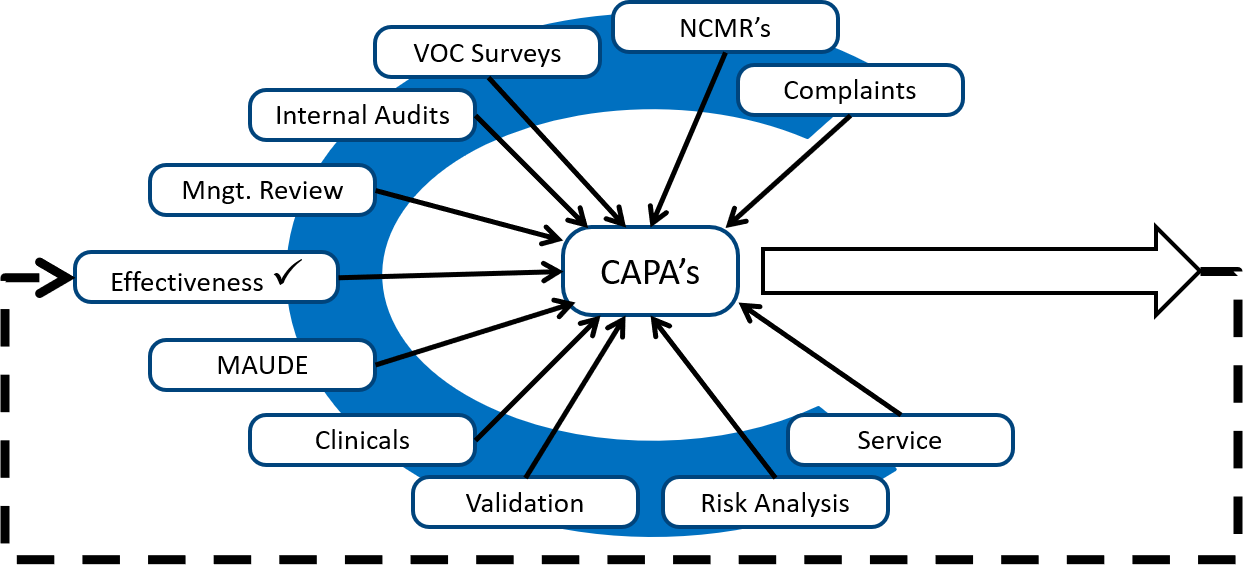
Monitoring and measuring each process is the best source of preventive actions, while internal audits are typically the best source of corrective actions. Any quality problems identified during validation are also excellent sources of corrective actions because the validation can be repeated as a method of demonstrating that the corrective actions are effective. However, your ISO 13485 certification auditor will focus on non-conforming products, complaints, and services as the most critical sources of corrective actions. These three sources are prioritized because these three sources have the greatest potential for resulting in serious injury, death, or recall if corrective actions are not implemented to prevent problems from recurring.
Step 4 – Conducting your first management review
In addition to completing a full quality system audit before your stage 1 audit, you are also expected to complete at least one management review. To make sure that you have inputs for each of the 12 requirements in the ISO 13485:2016 standard, it is recommended to conduct your management review only after you have completed your full quality system audit and initiated some corrective actions. If possible, you should also conduct supplier audits for any contract manufacturers or contract sterilizers. It is recommended to use a template for that management review that is organized in the order of the required inputs to ensure that none of the necessary inputs are skipped. Quality objectives will need to be established long before the management review so that the top management team has sufficient time to gather data regarding each of the quality objectives. Also, you should consider delegating the responsibility for creating the various slides for each input to different members of top management. This will ensure that everyone invited to the meeting is engaged in the process, and it will spread the workload for meeting preparation across multiple people.
At the end of the meeting, top management will need to create a list of action items to be completed before the next management review meeting. Meeting minutes will need to be documented for the meeting, including the list of action items and each of the four required outputs of the management review process. We recommend using the notes section of a presentation slide deck to document the meeting minutes related to each slide. Then the slide deck can be converted into notes pages and saved as a PDF. The PDF notes pages will be your final meeting minutes for the management review. An example of one of these note pages is provided in the figure below.

One of the more common non-value-added findings by auditors is when an auditor issues a nonconformity because you do not have your next internal audit and your next management review scheduled–even though each may have occurred only a month prior to the Stage 1 audit. Therefore, we recommend that you document your next 12-month cycle for internal audits and schedule your next management review as action items in every management review meeting. The schedule can be adjusted if needed, but this allows top management to emphasize various areas in internal audits that may need improvement. You might even set a quality objective to conduct a minimum of three management reviews per year at the end of your first management review.
Step 5 – Stage 1, Initial ISO 13485 Certification Audit
In 2006, the ISO 17021 Standard was introduced for assessing certification bodies. This is the standard that defines how certification bodies shall go about conducting your initial certification audit, annual surveillance of your quality system, and the re-certification of your quality system. In the past, certification bodies would typically conduct a “desktop” audit of your company before the on-site visit to make sure that you have all the required procedures. However, ISO 17021 requires that certification bodies conduct a Stage 1 audit that assesses the readiness of your company before conducting a Stage 2 audit. Therefore, even if the Stage 1 audit is conducted remotely, the certification body is expected to interview process owners and sample records to verify that the quality system has been implemented. Certification body auditors will also typically verify that your company has conducted a full quality system audit and at least one management review. Finally, the auditor will usually select a process such as corrective action and preventive action (CAPA) to make sure that you are identifying problems with the quality system and taking appropriate measures to address those problems.
Your goal for the Stage 1 audit should not be perfection. Instead, your focus is to make sure that there are no “major” non-conformities. The term “major” used to have a specific definition:
- Absence of a documented procedure or process
- Release of nonconforming product
- Repeat nonconformities (not possible during Stage 1)
Under the MDSAP, the grading system for nonconformities now uses a numbering system for grading nonconformities: “Nonconformity Grading System for Regulatory Purposes and Information Exchange Study Group 3 Final Document GHTF/SG3/N19:2012.” Any nonconformity is graded on a scale of one to four, and then two potential escalation rules are applied. If any nonconformities are graded as a four or a 5, then the auditor must assess whether a five-day notice to Regulatory Authorities is required. A five-day notice is required in either of the following situations: 1) one or more findings grading of “5”; or 2) three or more findings graded as “4.” If your Stage 1 audit results in a five-day notice, then you are not ready for your Stage 2 audit. For example, a complete absence of two required procedures in clauses 6.4 through 8.5 of ISO 13485:2016 would result in two findings with a grading of “4.” This would not result in a five-day notice, but the absence of a third required procedure would result in a five-day notice.
The duration of your Stage 1 audit will be one or two days, but a 1.5-day audit is quite common for MDSAP Stage 1 audits. The reason for the 1.5-day Stage 1 audit is that it is challenging to assess readiness for Stage 2 in one day, and if the total duration of Stage 1 and Stage 2 is 5.5 days, then the Stage 2 audit could be completed in four days. The four-day audit is more convenient than a three-day audit for a two-person audit team.
After your Stage 1 audit, you will receive an audit report, and you should expect findings. You should initiate corrective actions for each finding immediately, to make sure the findings are corrected and prevented from repeat occurrence before the Stage 2 audit. The duration between the audits is typically about 4-6 weeks. That does not leave much time for you to initiate a CAPA, perform an investigation of the root cause, and implement corrective action. At a minimum, you must submit a corrective action plan for each finding to your MDSAP auditing organization (AO) within 15 calendar days of receiving the finding. For any findings graded as a “4” or higher, you will need to provide evidence of implementing the corrective action plan to the AO within 30 calendar days of receiving the finding. You are also unlikely to have enough time to conduct an effectiveness check prior to the Stage 2 audit.
Step 6 – Stage 2, Initial ISO 13485 Certification Audit
The Stage 2 initial ISO 13485 certification audit will verify that all regulatory requirements have been met for any market you plan to distribute in. The auditor will complete an MDSAP checklist that includes all of the regulatory requirements for each of the countries that recognize MDSAP: 1) the USA, 2) Canada, 3) Brazil, 4) Austria, and 5) Japan. The auditor will also sample records from every process in your quality system to verify that the procedures and processes are fully implemented. This audit will typically be at least four days in duration unless multiple auditors are working in an audit team.
The audit objectives for the Stage 2 ISO 13485 certification audit specifically include evaluating the effectiveness of your quality system in the following areas:
- Applicable regulatory requirements
- Product and process-related technologies
- Technical documentation
All procedures will be reviewed for compliance with ISO 13485:2016 and the applicable regulations. The auditor will also sample records from each process. If the auditor identifies any nonconformities during the audit, it is important to record the findings and begin planning corrective actions immediately. If you have any questions regarding the expectations for the investigation of the root cause, corrections, corrective actions, and effectiveness checks, you should ask the auditor during the audit or the closing meeting. At a minimum, you must submit a corrective action plan for each finding to your MDSAP auditing organization (AO) within 15 calendar days of receiving the finding. For any findings graded as a “4” or higher, you will need to provide evidence of implementing the corrective action plan to the AO within 30 calendar days of receiving the finding. The auditor will not be able to recommend you for ISO 13485 certification until your corrective action plans are accepted.
If you receive a finding with a grading of “5,” or three or more findings graded as “4,” then the MDSAP auditor is required to issue a five-day notification to the regulators. The auditor will also need to return to your facility for a follow-up audit to close as many findings as they can. It is not necessary to eliminate all of the findings in order to be recommended for ISO 13485 certification, but the grading of the findings must be reduced to at least a “3” before recommending the company for certification. The number of findings also determines whether the auditor recommends your company for certification.
In addition to reviewing the findings and conclusions of the audit during the closing meeting, the auditor will also review the plan for the annual surveillance and re-certification with you. Each certification cycle is three years in duration. There will be two surveillance audits of approximately one-third of the duration of the combined duration of stage 1 and stage 2 initial certification audits, and the first surveillance audit must be completed within 12 months of the initial certification audit. In the third year, there will be a re-certification audit for two-thirds of the duration of the combined duration of stage 1 and stage 2 initial certification audits. The initial ISO 13485 certificate will be issued with a three-year expiration, and the certificate is typically received about one month after the acceptance of your corrective action plan.
Q&A
There are no stupid questions, and we can save you weeks of wasted time if you just ask for help. We are always looking for new ideas for blogs, webinars, and videos on our YouTube channel. If you have any general questions about obtaining ISO 13485:2016 certification, please email Rob Packard at rob@fdaestar.com. If you have a suggestion for new ISO 13485 training materials, you can also use our “Suggestion Portal.” You can also schedule an initial free consultation with Rob using his calendly link.
About Your Instructor
 Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.
Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.
Thank you for all your work on our behalf…
We are looking to have our 13485 certification with a US based company and would like to have your recommendations. I have already made contact with Eagle based on your recommendation and would like a couple of others.
Thank you,
Jim McGhan
There are essentially 3 tiers of certification bodies. The first tier is an MDSAP auditing organization. They are the most expensive. The second tier is a Notified Body that is not an MDSAP auditing organization. The third tier is a certification body that can do neither MDSAP nor CE Marking. Eagle is the top of the third tier. If you don’t need a first tier or second tier certification body, I would not bother with a second quote.
Thank you for arranging the platform to gain knowledge and experience.
You’re very welcome.