Are you a start-up device company that needs medical device consulting services and training for an FDA 510k submission?
How much does a 510(k) cost?
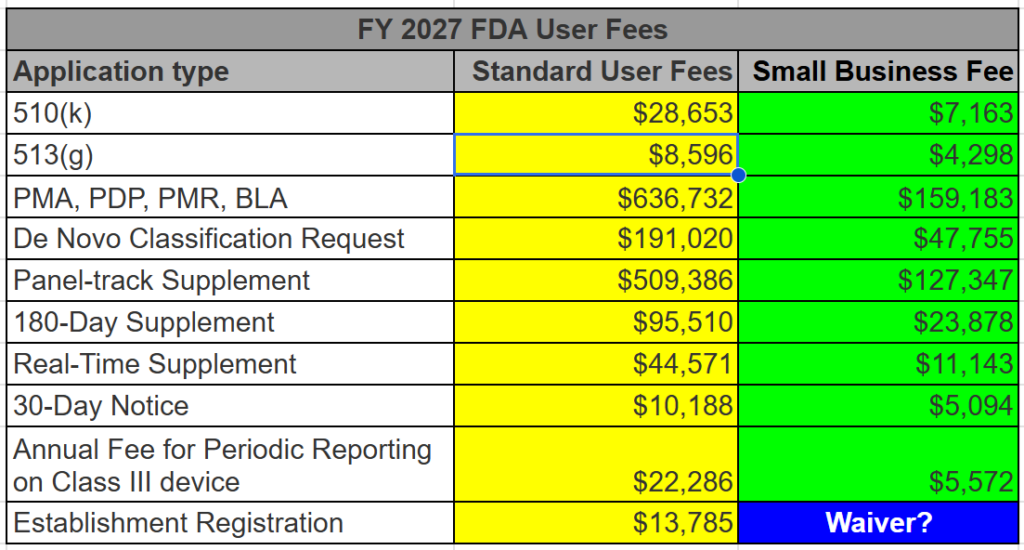
The FDA user fees for medical device 510k submissions, 513(g) submissions, and De Novo Classification Requests are all discounted for qualified small businesses. To qualify, you must submit a small business qualification form (FDA Form 3602N) each new fiscal year (October 1 – September 30). We recommend that every company submit on August 1 for the next fiscal year–even if they are unsure if they will submit. This advice alone will save your firm thousands of dollars. Below is a table listing all of the FDA user fees for submissions–both standard and small business fees.
Our vision
We are trying to help MedTech companies successfully develop devices that are safer and more effective. Individually, we might develop a single medical device and become wealthy. As medical device consultants helping others, we can help 100 companies every year. In 20 years, our clients will develop more than 2,000 new medical devices. That can change the world.
Medical device consulting services
Medical Device Academy, Inc. is a quality and regulatory consulting firm. We have ten (10) people on our team, and everyone works virtually from home. We specialize in helping small device companies prepare FDA 510k submissions using the eSTAR template, preparing FDA pre-submissions using the PreSTAR template, implementing new quality systems for compliance with the FDA, ISO 13485, and MDSAP, and conducting quality system audits. We also help with FDA US Agent services, CE Marking preparation, and Canadian License applications. Clients with urgent needs where time to market is critical turn to us. Our passion is teaching medical device professionals how to prepare for future regulations. For more information, please visit our website or our YouTube channel. We even wrote a 510k book, “How to prepare your 510k in 100 days,” because 100 days is how long your testing will take. It is only available as part of our 510k course series, and you can find all the details on our 510k course webpage.
Buy the 510(k) course series for $1,495

What do we charge for medical device consulting to prepare an FDA 510k submission?
The estimated cost of preparing a medical device 510k submission is $17,500. This pricing does not include pre-submission meetings or the FDA user fees for FY 2026. A table with user fees is found below as well. A button for our standard pricing sheet of consulting services is provided below, along with a second button for our turnkey quality management system (QMS) pricing.

How long does it take to obtain a medical device 510k clearance?
Overall, the average is 125 days, but you can only estimate the time to obtain medical device 510k clearance by analyzing the data from recent, previous submissions for the same product classification. Every classification is different, and the average for each product classification ranges from less than 90 days to more than 180 days. We can explain the various options for reducing the review time (e.g., third-party reviewers), but the biggest delays are caused by the need to repeat testing because clients skipped a pre-submission meeting.
Medical device product classifications and regulatory pathways
The FDA regulatory pathway and other regulatory pathways are dependent upon the classification of the device. We provide regulatory pathway analysis services to clients for the US market, the European market, and the Canadian market. We also help you determine the testing requirements, identify potential predicates, and explain the quality system requirements for your type of device. This is typically included with our medical device 510k pre-submissions and De Novo Applications.
FDA substantial equivalence (SE) decision process
The process flow diagram in the video above may seem simple, but how you interpret each question can change the answer significantly. Sometimes, a few words or a single design feature can change the FDA reviewer’s decision when evaluating substantial equivalence. Many of these differences concerning interpretation become evident during the pre-submission meeting with the FDA. Following the wrong path will result in significant delays at best, and many clients discover that clinical testing and/or a De Novo Application is required due to changes in technological characteristics or due to a slight change in the indications for use.
Developing a verification and validation (V&V) testing plan
Having the right verification and validation testing plan is critical to timely FDA 510k clearance and approval of De Novo Requests. That’s why we prepare draft testing plans for clients, and then we ask the FDA for feedback on the testing plan during a pre-submission meeting. We use guidance documents, competitor submissions, and our experience with similar devices. However, we often learn something new from the FDA at every pre-submission meeting because the requirements are constantly changing.
Medical device 510k project management
Most people see meetings as a waste of time, but we try to conduct efficient, weekly, 30-minute meetings with medical device consulting clients. The purpose is to answer their questions, give them an update on the project status, and make sure there are no surprises that could delay the project’s completion. Typically, we are waiting for the final testing reports, such as 1) the sterilization validation report, 2) the sensitization testing report, or 3) the electrical safety testing report. For every project, we use the table of contents as a color-coded planning tool to track the status of every document needed for your submission.