There are four differences between a De Novo application and a 510k submission, not including time or money.
The best regulatory experts plan regulatory submissions months before the performance testing is completed, and often the strategic regulatory pathway is determined before the design of the device even begins. If your design team is developing innovative technology, you may have difficulty finding a predicate device that is substantially equivalent to your device. If you have not completed a De Novo application before, where do you start?
History of the De Novo
Historically, a De Novo application required that your device be submitted through the 510k process first. If the FDA determined that your device was not substantially equivalent to the predicate you chose, then you received a “Not Substantially Equivalent” (NSE) letter from the FDA. Once you receive an NSE letter, you have three options: 1) select a different predicate and re-submit, 2) re-submit the device through the Pre-Market Approval (PMA) process, or 3) submit the device through the De Novo application process. You could not submit a De Novo application until you received an NSE letter.
The De Novo application process was revised on July 9, 2012 to allow manufacturers to submit a De Novo application without a preceding 510k submission. This was done because many products are technologically equivalent to a predicate device, but the indications for use are quite different. For example, many in vitro diagnostic (IVD) products are indicated for diagnosing new viruses, but the device uses technology equivalent to another IVD product the manufacturer already makes. For this reason, most of the first 100+ De Novo application approvals were for IVD products.
Guidance document
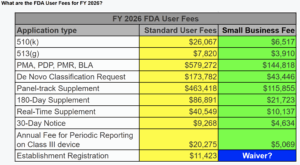
The De Novo application process finally has an approved final rule and 2021 FDA guidance document, and the fees charged for De Novo applications as part of MDUFA V for FDA user fees, are significantly higher than 510k submissions.
Using Q-sub meetings
Pre-sub meetings are generally recommended by the FDA for manufacturers that intend to submit a De Novo application without a prior 510k submission and subsequent NSE letter. If the device is a Class II, a pre-sub meeting allows the manufacturer to request input from the FDA, regarding performance testing and special controls. The draft De Novo guidance document specifically recommends following the existing content guidelines for a pre-sub meeting request, but the guidance also recommends including the following elements in the pre-sub meeting request:
- Proposed classification rationale (i.e., Class I, Class II exempt, or Class II)
- Details of efforts previously taken to identify a predicate
- Risks and benefit-risk analysis
- Proposed FDA special controls
Before you submit a pre-submission request for a potential De Novo application, you should consider the following questions. First, is your device suitable for a De Novo application (see point #2 above)? Second, when is the ideal time for you to submit your application? At least 90 days before your design freeze is needed if the pre-submission meeting request is going to have any impact on the performance testing plans. Third, what questions do you want the FDA to answer regarding data requirements? Sometimes, providing preliminary data can help you persuade the FDA to accept less total data for approval of the final application. Finally, you may want to consider preparing a draft list of FDA special controls for the FDA to review as an attachment to your FDA PreSTAR.
A De Novo for Class I and Class II exempt devices?
Most manufacturers mistakenly assume that De Novo applications are only for devices that are Class II and will require a 510k submission for future product submissions in the same classification. However, the regulations require that the application cover letter include both a “Classification Summary” and a “Classification Recommendation.” The recommendation for classification may be for Class I, Class II exempt, or Class II non-exempt. If you recommend that the FDA classify the device as Class II exempt, then the recommendation must include a justification for why premarket notification is not required.
Regardless of which classification is recommended, the classification justification must be based on a benefit-risk analysis. Class I and Class II exempt classifications are likely to be recommended more in the future for many of the standalone software products that are being developed by manufacturers because those software devices generally have a low risk. Existing product classifications may be used, but if the indications for use are not substantially equivalent to a predicate, the manufacturers will submit a 510k and receive NSE letters. For the companies that are claiming substantial equivalence to products that already have a product classification that is exempt from premarket notification, manufacturers will continue to register and list products under existing classification codes until the FDA intervenes–even if the indications for use are not equivalent.
How are 510k and De Novo content different?
For a De Novo application, a large percentage of the sections required for a 510k submission are the same. The FDA guidance identifies one unique section of a De Novo: the cover letter (i.e., Attachment II in the De Novo guidance). However, there are two sections of a 510k submission that also need to be eliminated for a De Novo application:
- 510k Summary or 510k Statement is not required, because this is not a 510k submission
- Substantial Equivalence Comparison, because a De Novo does not claim equivalence to a predicate
Do you need help submitting your De Novo Classification Request?
De Novo Classification Requests can still be submitted as an FDA eCopy, but we no longer offer FDA eCopy print and ship services. Since July 2022, manufacturers can upload an FDA eCopy to the Customer Collaboration Portal (CCP). However, the FDA also permits using the new FDA eSTAR templates to submit a De Novo. This is a much better way of organizing your submission because the templates automatically verify completeness, include help and links to guidance documents throughout the entire template, allow you to submit non-PDF document formats (including larger files), and the submission process is much easier–you upload just one PDF file, and there is no need to zip any files or worry about file name formats.
New related webinar
Companies developing devices with truly innovative technologies frequently have difficulty identifying suitable predicate devices. The best regulatory experts plan in advance for these regulatory submissions by honing their knowledge of the De Novo application process. On Thursday, August 1, 2024, we recorded a new webinar sharing our tips and templates for De Novo applications. Click here to learn more about the webinar.