This article explains how to write your classification recommendation for a De Novo Classification Request using a risk-based approach.
“Automatic Class III Designation” does not mean that your device is a Class III device. That phrase means that the device is new, and therefore it will be automatically classified as Class III until a company submits a De Novo Classification Request. You and your company, not the FDA, should make the classification recommendation and propose the regulatory pathway for a new device. Submitting a 513g request is an option, but a 513g request involves paying the FDA money to write a classification recommendation. The FDA will always be more conservative in their assessment than the manufacturer.

Although no FDA guidance explains how to write a classification recommendation, companies have been writing these documents for years–for Technical Files. Most countries have risk-based classification rules, while the FDA’s product classification database is centered upon precedents and adjusted over time by historical trends of adverse events and recalls. Therefore, you should write a classification recommendation for the FDA that is focused on a documented risk assessment. Your approach will also need to be modified to include classification information for similar indications for use and technological characteristics that are already established in the US market.
Most Common Mistakes in Writing a Classification Rationale
Many people mistakenly write a short classification rationale for a technical file, which simply states which classification rule applies and why. Although this approach is acceptable for a Declaration of Conformity, you must provide a comprehensive classification rationale in your technical file. First, you need to make sure that there is only one classification rule that applies. For example, classification rules fall into four general categories:
- Non-invasive Devices
- Invasive Devices
- Active Devices
- Special Rules
The software was haphazardly added to the active devices category until recently, and special rules were created to address emerging areas of interest and concern. Therefore, most active devices have a second rule that applies regarding the invasive nature of the device–or lack thereof. In order to write a comprehensive classification rationale, you need to review each classification rule and document your explanation for why it applies or does not apply to your device.
A Classification Recommendation Compares Indications for Use
The FDA does have classification rules, but the rules are not 13 numbered items in the Code of Federal Regulations (CFR). The FDA expects a risk assessment of comparing your device with existing devices on the US market. The basis of comparison should be: 1) the indications for use and 2) the technological characteristics. First, you should identify other devices that have similar indications for use. For example, a device intended for home use or over-the-counter (OTC) use represents a higher risk to patients and users than a device intended for prescription use only. Patients may fail to identify contraindications for a device properly, or the lack of formal medical training may result in use errors that would not occur when a physician uses the same device.
Other aspects of indications for use that impact the risk assessment are the part of the body where your device will be used and the duration of use. For example, implants are at higher risk than non-implants, because implants are in contact with the body for a much longer period of time. Implants can also expose the body to systemic risks, while a surface contacting device is likely only to have a localized effect. Degradation of implants also exposes the body to small particles, with more surface area, that can travel from one part of the body to another.
If your device is used for life support, the device will also be considered at higher risk than devices that are not required for life support. If your device is the only device used for diagnosis, this also represents a higher risk than a device that acts as an adjunct to other devices. Finally, if your device is an accessory to other devices that are high risk, your device may be considered a higher risk as well–especially if it controls the higher risk device.
In your analysis, you need to identify devices that are already on the US market that have similar indications for use. Usually, those devices will be Class II devices. However, if some of those devices are Class I or Class III, you will need to be more careful with how you differentiate your indications for use from those other devices.
A Classification Recommendation Compares Technological Characteristics
When comparing technological characteristics, the following aspects should be considered: 1) materials, 2) design, 3) energy source, and 4) other design features. For example, absorbable materials are generally considered at higher risk than devices that are not absorbable. Sterile devices are generally at higher risk than non-sterile devices because the failure of the sterilization process or the package integrity can result in serious infections and death. Devices that are electrically powered are usually considered at higher risk than devices that are not powered. Finally, software-controlled devices that provide feedback control are considered at higher risk than a device that does not have feedback control. Each technological characteristic also represents a different category of hazard. Hazard categories are listed in Table C1 of Annex C in ISO 14971:2019. These include chemical, biological, electrical, radiation, etc.
Once you have identified the Classification of other devices with similar indications for use and technological characteristics, you need to estimate the risks for each hazard identified. This involves more than just listing hazards and assigning scores for severity and probability for the occurrence of harm. Severity should consider the type of injuries, the number of injuries, and the duration of harm. Probability should consider the frequency of events (P1), and the probability of events resulting in injury (P2). These risk estimates also require clinical data.
Benefit-Risk Analysis
In the end, you prepare a benefit-risk analysis for your device. This is much more than a statement that the benefits outweigh the risks. You need to identify the clinical benefits of your device when compared to alternative treatments. You also need to analyze risks relative to alternative treatments. You will need to prepare this as a summary of risks–not a list of hazards. Ultimately, your benefits should be equivalent to the benefits of existing devices on the market or better, and the risks should be equivalent to existing devices on the market or less.
Examples of Classification Recommendation
Eight different medical devices are legally marketed in the USA for weight loss or weight management:
- Lap-Band Adjustable Gastric Banding System – Class III, PMA
- Maestro Rechargeable System – Class III, PMA
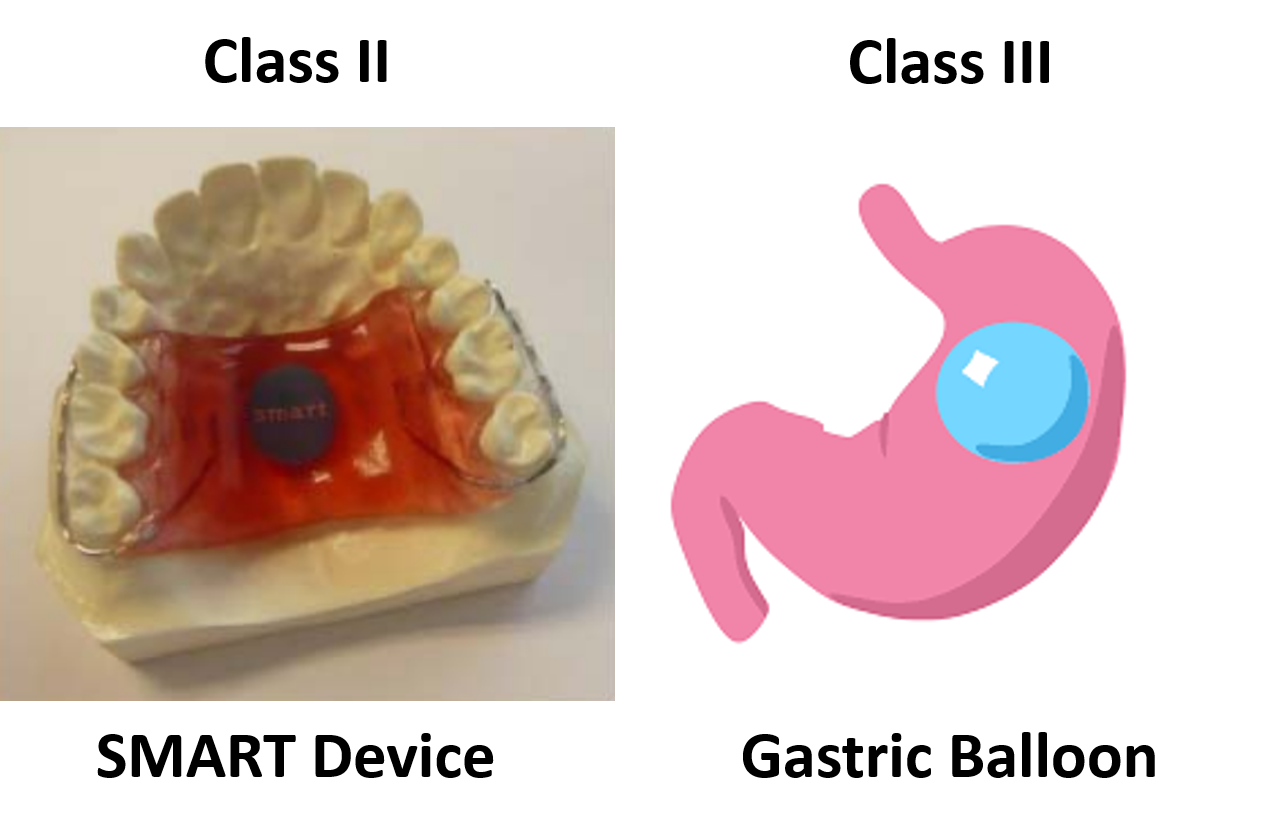
- ORBERA Intragastric Balloon System – Class III, PMA
- Obalon Balloon System – Class III, PMA
- TransPyloric Shuttle/TransPyloric Shuttle Delivery Device – Class III, PMA
- AspireAssist – Class III, PMA
- Sensor Monitored Alimentary Restriction Therapy (SMART) Device – Class II, De Novo
- Plenity – Class II, De Novo
The indications for use for these products are similar, but not identical. Plenity is indicated for patients with a BMI of 25 – 40 kg/m2. In comparison, ORBERA is indicated for patients with a BMI of 30-40 kg/m2, and AspireAssist is indicated for patients with a BMI of 35-55 kg/m2. All three of these indications have overlapping BMI ranges. However, the clinical benefits to a person with a BMI of 25 kg/m2 are not the same as the clinical benefits to a person with a BMI of 40 or 50 kg/m2. Therefore, these minor differences in BMI can have a significant impact on the benefit/risk analysis used for a De Novo approval decision and the Classification (i.e., Class II or Class III) determined by the FDA.
The only two weight management devices that received the approval of the De Novo Classification Request had very different technological characteristics from the other six devices. All six Class III, PMA devices, are implants, while the Class II devices are not implants. The risks associated with implants are much greater than with non-implants. The risk of implants breaking or leaking, and the difficulty in removing an implant, are just two of the considerations that must be evaluated in deciding whether an implantable device should be a Class II or Class III device.
Pingback: De Novo Application - If there is no 510(k) predicate Medical Device Academy