Today the FDA released a press release announcing plans to implement an alternate 510k pathway called the “Safety and Performance Based Pathway.”

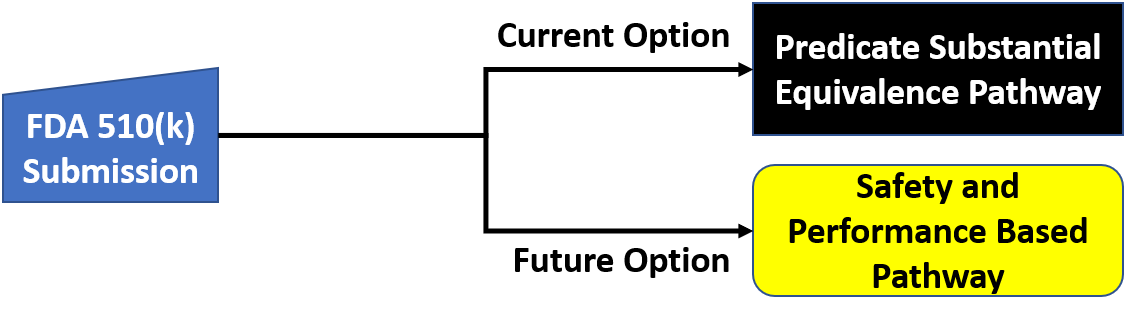
What is the current 510k pathway for clearance of medical devices?
The current version of the 510k pathway is defined in a guidance document on a substantial equivalence that was released on July 28, 2014. The pathway involves six questions that an FDA reviewer must answer before it can be determined whether a new device is equivalent to an existing device that is legally marketed in the USA. These are the six questions:
- Is the predicate device legally marketed?
- Do the devices have the same intended use?
- Do the devices have the same technological characteristics?
- Do different technological characteristics raise different questions of safety and effectiveness?
- Are the methods of evaluating new/different characteristics acceptable?
- Does the data demonstrate substantial equivalence?
Five (5) ways the FDA strengthened the current 510k pathway
Today the FDA released an 8-page presentation summarizing five (5) ways that the FDA strengthened the current 510k pathway during the past several years. The five ways are:
- Increased expectations for the content of a 510k submission
- Implementation of the refusal to Accept (RTA) policy
- Improved consistency and thoroughness of the 510k review process
- Elimination of the 510k pathway for Class III devices
- Eliminated the use of > 1,000 unsafe devices as legal predicates
You may have been complaining that 510k requirements seem to change constantly. Now you have proof that the changes to the 510k pathway are part of a strategic plan implemented over the past decade. Lawyers may argue that the resulting regulations go well beyond the intent of the original 510k legislation. This is completely true. The cumulative effect of implementing dozens of 510k guidance documents is that the official interpretation of the 510k section of the Food and Drug Act now has little resemblance to the original legal intent.
The original intent of the 510k legislation was to allow competitors to copy an existing device that is legally marketed in the USA. Cumulative changes to a device that existed in 1976, eventually result in a completely new device. The word “equivalent” has been perverted to such an extent that thousands of devices now exist that do not even remotely resemble devices from 1976. The FDA recognized this around 2007, and the US device regulations began to “strengthen.”
What is the basis for the Alternate 510k Pathway?
The basis for the alternate 510k pathway is the submission of data that is safety and performance-based instead of comparison to an older predicate. In addition, the new pathway will enable you to make comparative claims by demonstrating that the new subject device meets or exceeds the safety and performance criteria. There is also a goal to use the pathway as a potential method of harmonizing the US medical device regulatory process with other global medical device regulations. The new process, combined with improved post-market surveillance, will complement the FDA’s work on NEST by allowing the FDA to rapidly require the implementation of risk controls to address identified safety issues.
What is the expected timeline for the implementation of the Alternate 510k Pathway?
The alternate 510k pathway has been in development for quite some time. Jeff Shuren first announced the plan to create the alternate 510k pathway at AdvaMed’s MedTech conference in San Jose, California, in September 2017. On Monday, December 11, 2017, the FDA announced that draft guidance would be released in Q1 of 2018. On April 12, 2018, the FDA finally released the draft guidance for public comment.
The FDA intends to release final guidance for the new alternate 510k pathway in early 2019. This pathway will initially be limited to “well-understood device types”–probably as a 510k pilot program. You can expect this new pathway to be released in a similar way to the Special 510k expansion pilot and the Quik 510k pilot. That final guidance will be released, and the pilot will begin immediately after the release of the guidance.
Is this new process likely to require significant changes to future 510k submissions?
The phrase “significant changes” is subjective, but if you look at the current 20 required sections of a 510(k) submission, there is only one section that would be required to change for the new alternate 510k pathway. Specifically, section 12 is currently used for a substantial equivalence comparison. This section would not be applicable under the alternate 510k pathway. Under the alternate 510k pathway, you can expect the FDA to require at least a summary of the safety and performance data to be submitted for approval of the subject device.
Another change you can expect is that all devices submitted under the alternate 510k pathway will be required to have a benefit-risk analysis in accordance with the corresponding FDA guidance. This new guidance was released on September 25, 2018, as a draft. However, a benefit-risk analysis is required for De Novo applications, CE Marking applications, and, logically, the FDA will also require this for 510k submissions that do not rely upon equivalence to the predicate device.
More Information on the Medical Device Safety Action Plan
The FDA created a webpage on its site, providing information about the Medical Device Safety Action Plan. The page includes several hyperlinks to documents with more information. Below are a few of the relevant links:
- Link for Downloading the Medical Device Safety Action Plan
- FDA Commissioner’s Statement on Modernizing 510(k) Program
- FDA Has Taken Steps to Strengthen the 510(k) Program – November 26, 2018
The FDA also indicated that a new guidance for De Novo applications would be released in a couple of weeks. Please subscribe to our blog, and you will receive notification of a blog in response to that guidance when it is released.
Great stuff ! Thanks &Thumbs up for posting this.
Keep it up!
PS I’d like to suggest another complimentary topics
https://axisbits.com/blog/Modern-Medical-App-Development-Checklist
https://www.slideshare.net/G3Com/how-to-create-modernized-mobile-experiences?qid=59988110-eb65-4a8f-8132-fadfd4ac1241&v=&b=&from_search=10
https://www.forbes.com/sites/yiannismouratidis/2018/12/09/the-rise-of-use-of-medical-devices-force-fda-to-change-the-rules/#26d015d32101
Thank you for the great suggestions for related links.
Pingback: Regulatory pathway analysis--a case study Medical Device Academy