In this use error training webinar, you will learn how to use our decision tree form to determine if you have identified a use error or an abnormal use.
Your cart is empty
Use error and abnormal use training webinar ($79)
In this webinar, you will learn what a use error is and what abnormal use is. You will learn how to use our decision tree form to determine if you have identified a use error or an abnormal use. The webinar is a 21-minute recorded video (i.e., mp4) and includes the decision tree form with a work instruction.
Use error and abnormal use training webinar
In this webinar, you will learn what a use error is and what abnormal use is. You will learn how to use our decision tree form to determine if you have identified a use error or an abnormal use. The webinar is a 21-minute recorded video (i.e., mp4) and includes the decision tree form with a work instruction.
Price: $79.00
Please note: These products will be delivered to the email address provided in the shopping cart transaction. After the transaction is verified, please check your email for the download.
When is the use error and abnormal use training webinar?
This webinar is a 21-minute recording you can purchase on-demand and watch the training as often as you wish. If you need help preparing a usability engineering file (UEF) for your device, we can help you on an hourly basis. Please contact Lindsey Walker for a quote.
What you will receive in the Use error and abnormal use training webinar:
Other Usability Engineering / Human Factors Training
Matthew Walker – QMS, Risk Management, Usability Testing, Cybersecurity
Matthew brings a unique background as a former Firefighter/EMT and Rope Rescue Tech with experience in OSHA and NFPA regulations. For the better part of a decade, he has worked as a Technical/Medical Writer and Lead Auditor. He holds degrees in Fire Science and Computer Forensics and Digital Investigations, graduating Summa Cum Laude from Champlain College. Matthew is also an active member of several academic honor societies including Omicron Sigma Sigma’s Order of the Sword and Shield. His professional focus includes Human Factors Engineering, Risk Management, and Cybersecurity with a special interest in applying Digital Forensics and Incident Response (DFIR) practices to medical technology. He combines regulatory expertise with technical insige to strengthen both product safety and oranizational resiliance.
The FDA and Health Canada both have executive-level orders requiring medical device shortage reporting or supply-chain disruptions.
In a previous article, we discussed supply-chain disruptions and mentioned that there might be medical device shortage reporting requirements if that disruption causes a market shortage of the manufactured device. Both the United States and Canada have reporting requirements for supply disruptions or the market’s ability to meet the demand of specific types of devices.
Both the U.S. FDA and Health Canada have executive-level orders that require reporting of shortages or disruptions to the supply of medical devices deemed necessary for the COVID-19 Health Emergency. There is some overlap, but each country is monitoring and experiencing shortages and disruptions of different devices.
Where did medical device shortage reporting responsibilities come from?
Check 21 CFR 820, ISO 13485:2016, and even peek at SOR 98-282 and see if you can find your obligations for reporting. Go ahead. I’ll wait… Not much in there, right? Adverse events, complaints, etc., but not market shortages. Medical device shortage reporting is specific to health emergencies. The U.S. FDA and Health Canada happen to be two authorities having jurisdiction with reporting requirements for shortages concerning the COVID-19 Health Emergency. However, there may be others, so having your organization’s regulatory affairs manager verify the reporting requirements for the markets in which you are engaged might not be bad.
U.S. FDA 506J reporting-
U.S. FDA logo
In the United States, an Amendment to the U.S. Food, Drug, and Cosmetics Act requires regulatory reporting by medical device manufacturers to the U.S. FDA. It is sometimes called 506J reporting for the Section of the U.S. FD&C Act where it is located.
You will find the statutory requirements outlined within 21 USC 356J.
21 USC 356J Discontinuance or interruption in the production of medical devices
For the full text read, 21 USC 356j: Discontinuance or interruption in the production of medical devices. (Interestingly enough, the website where this information is available is not an HTTPS site, so visit at your own discretion).
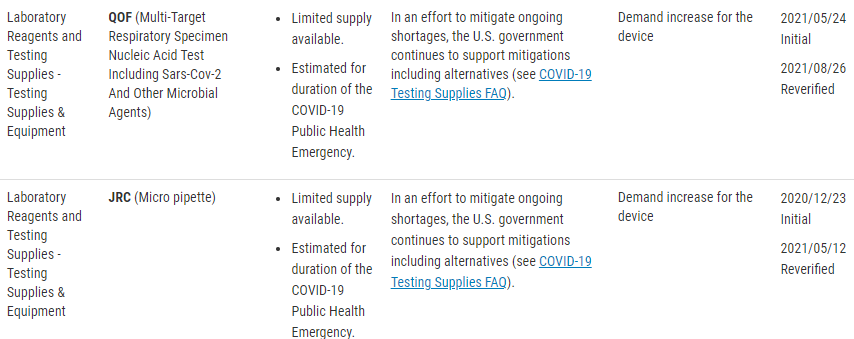
There are two types of devices that the FDA is monitoring. “Critical” devices and an FDA-published list of devices for which COVID-19 is causing a higher than expected demand.
The FDA has released a guidance document that contains criteria for what is considered to be a “Critical Device”. This includes devices such as those used during surgery, emergency medical care, and those intended to treat, diagnose, prevent, or mitigate COVID-19.
Screenshot of the Critical Device Criteria for 506J reporting
There is also a published list of concerned devices that the FDA is specifically monitoring. The FDA website lists these devices by product code, but include the following device types;
Clinical Chemistry Products
Dialysis-Related Products
General ICU/Hospital Products
Hematology Products
Infusion Pumps and Related Accessories
Microbiology Products
Needles and Syringes
Personal Protective Equipment (PPE)
Sterilization Products
Testing Supplies and Equipment
Ventilation-Related Products
Vital Sign Monitoring
Screenshot of the FDA Shortage List
Understandably this process may not be intuitive, and for this, the FDA has released a guidance document that addresses;
Who must make the notification
When you should make a notification
What information needs to be included within your 506J notification
How to make a notification, and
Penalties for failure to make a notification
The referenced product codes may not be an all-inclusive list or entirely up to date. The best suggestion for full compliance is to go straight to the source of the regulation, in part because noncompliance can result in enforcement action from the FDA. If you think that your device might require notification to the FDA but isn’t in the reference table, you should contact the FDA for notification clarification. Below is the quote from the FDA website, and it includes the contact email for asking these specific questions to ‘the agency.’
“If a device type is not included in this table, but you believe it requires a notification under section 506J of the FD&C Act, or if you have questions regarding the device types in this table, you should contact FDA at CDRHManufacturerShortage@fda.hhs.gov and include “Question” in the subject line of the email.”
Link to the FDA Guidance Document for 506J Reporting- HERE
How to make a 506J report to the U.S. FDA?
The FDA accepts 506J reports in multiple ways. For example, you may use the 506J Reporting web form or submit a notification by email directly to (Include Email Here). In addition, Medical Device Academy has developed a Work Instruction and Form to determine if your company is experiencing a reportable discontinuance or meaningful disruption in manufacturing a medical device as well as compiling the report for submission.
There are a few methods of notification, a web form for individual notifications and spreadsheet options for multiple notifications at once, or emailing a report directly to the FDA reporting email included below;
CDRHManufacturerShortage@fda.hhs.gov
Screenshot of the FDA 506J reporting Webforms from https://fdaprod.force.com/shortages
It is for this process that Medical Device Academy developed WI-010 506J Shortage Reporting to the U.S. FDA. This work instruction and associated form, FRM-053 506J Reporting Form are designed to walk you through the process of determining reportability and compiling the information necessary to either complete the webform or email the report directly to the shortage reporting email.
Medical Device Shortage Reporting to Health Canada
Health Canada logo
Rather than discontinuance and disruption of manufacture, Health Canada is monitoring for shortages of specific devices. Therefore, Health Canada wants Medical Device Shortage Reports regardless of the reason for the shortage. It also shows that this is not identical reporting of the same conditions to two different authorities. Health Canada will also accept reports from Importers because the frame of reference is Canada’s supply of medical devices concerning Canada’s needs.
As an Authority Having Jurisdiction, Health Canada also has reporting requirements for medical device shortage reporting of specific types of medical devices. Health Canada is also an independent authority that uses a different device classification system than the U.S. FDA.
The table below shows the device types by their classification level that HC requires supply chain disruption notifications for. This information is current as of September 5th, 2021, and the link below will take you to the HC website page for the most up-to-date list.
List of ‘Specified Devices’ that Health Canada is monitoring for shortage reporting
One of the things that Health Canada does an excellent job of is defining its expectations. In the Second Interim Order Respecting Drugs, Medical Devices and Foods for a Special Dietary Purpose in Relation to COVID-19, it is explained the Manufacturers or Importers should report to the Minister actual or expected shortages of the device, OR components, accessories, or parts. These notifications must be made within 5-days of becoming aware of the shortage or the anticipated shortage date. Update reports must be made within 2-days of becoming aware of new information regarding the shortage, and a closing report must be made within 2-days of the end of the shortage.
(This link is to the HC website for the 2nd Interim Order referenced above)
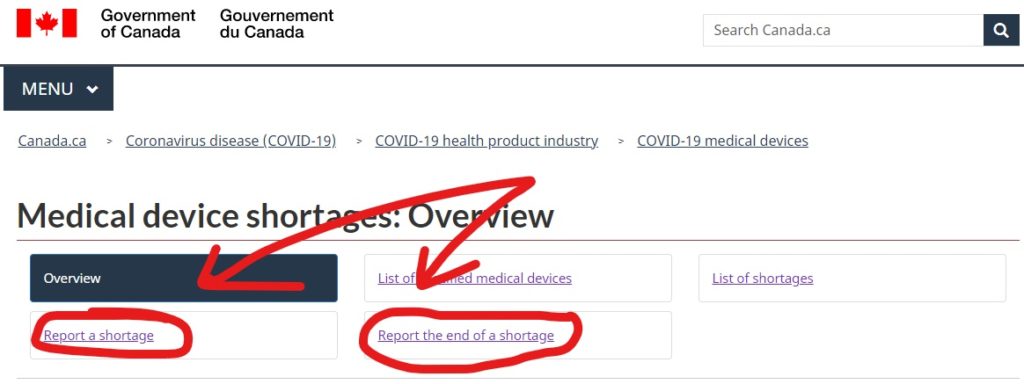
These reports are submitted online through the Health Canada Website. They have an entire section dedicated to medical device shortages, and the reporting links can be found there (Link here). If you have any questions or are on the fence about notification, you can email Health Canada at MD.shortages-penurie.de.IM@canada.ca.
Health Canada Webforms for reporting a shortage and the end of a shortage
The webform for reporting a shortage is the same webform that is used for providing update reports to Health Canada as well. This is both for manufacturers of specified medical devices as well as importers.
A profound realization was made while performing a routine MDR gap analysis of Medical Device Academy’s technical documentation procedure.
In this article I wanted to discuss the functional effect that a gap analysis can have on your entire quality system. Everything mentioned below is because I performed a MDR gap analysis against a single procedure which resulted in the addition of three words to a single sentence. This small modification was made simply for clarification of a sentence that was already compliant without the change. Those three words made me reexamine the entire procedure. Then I tried to identify possible interpretations of that one sentence both before and after the modification. Finally, I questioned how adding three words might affect quality systems as a whole.
What was the section reviewed in the MDR gap analysis?
The MDD (i.e. 93/42/EEC) did not include a section that defined the requirements for technical documentation. The MDD does not include the phrase “device description,” or “intended patient population.” Therefore, when the MDR came into force, companies were forced to update their technical documentation procedure to comply with the new Annex. The section of the regulation that I was performing the MDR gap analysis against was Annex II. Specifically, subsections 1.1a) and 1.1c):
1.1(a) “product or trade name and a general description of the device including its intended purpose and intended users“;
1.1c) “the intended patient population and medical conditions to be diagnosed, treated and/or monitored and other considerations such as patient selection criteria, indications, contra-indications, warnings;“
There are only two places in the MDD where the phrase “intended users” is found: Article 11(14) and Annex I(1). In Annex I(1) of the MDD, the Directive clarified that design of devices shall include: “consideration of the technical knowledge, experience, education and training and where applicable the medical and physical conditions of intended users (design for lay, professional, disabled or other
users).” The introduction of the phrase “intended patient population” in the MDR forced me to reevaluate the wording we were using in our SYS-025 Technical Documentation Procedure. The wording we were using was: “users and patients.” Therefore, first I added the word “intended” before “users” and “patient”, and second I added the word “population” after “patient.”
Why would the MDR require these specific changes?
These are very small changes but the changes were meant to more clearly explain that documentation was needed for very specific areas. Previous versions of the procedure left more room for interpretation that intended users may not have been differentiated as strongly from intended patients, especially for cases where they are one in the same. These two subsections of Annex II, 1.1 (a) and 1.1 (c), outline that there are two specific populations of real people that must be taken into account within the device description and design specification areas of your technical documentation:
the intended users, and
the intended patient.
Even if the user and the patient represent the same person, these are two separate areas that require technical documentation. Intended users, whom may or may not be within the “intended patient population” that the device was designed for, should be entirely separate on your technical documentation.
Take for example, a home use lancet device included within a glucometer kit. The intended user is probably going to be the diabetic patient who wishes to check their blood glucose levels at home. In this case the intended user would also be a member of the intended patient population.
However, because this is not always the case there should be a clear separation of the documentation between the intended users of 1.1(a) and the intended patient population in 1.1(c). An example of this would be something like a surgical scalpel. A medical device that would probably be intended to be used by a physician within the controlled environment of a surgical procedure. In this example scenario the intended patient population would differ from the user because the patient would be the population of people who would need to undergo the above mentioned surgical procedures, but the user of the device is the physician or surgeon actually performing the procedure.
Considerations going beyond my MDR gap analysis
Everything that we are talking about is for intended patient populations or intended users. Documentation regarding these areas is important for several reasons and strong record keeping early on in the device development stages will help with things like statistical analysis, tracking and trending, and even possible modifications to Instructions For Use or labeling in the future. Most people performing a gap analysis would just make the changes and move forward without a second thought. However, the phrase “intended patient population” was introduced to the MDR for a reason, and it forced me to think beyond the task at hand.
Let us look back at our diabetic patient with the home use glucometer kit. I like fleshing my characters out, and providing a back story really helps me mentally associate these fictitious characters with the potential real-life patients they may represent.
I am going to name him Matthew D. Mellitus Jr. He is 28 years old. A morbidly obese type II diabetic, and a married father of two. Beyond the extraordinary play on words with Mr. D. Mellitus, II is I promise that there is a purpose behind this.
Matt is the intended user of the specific glucometer kit that he has. It contains within it, a glucometer, alcohol prep pads, a lancet device, spare lancets, and a container of test strips. He is also a member of the intended patient population because he is a diabetic with orders from his primary care physician to check his blood glucose levels at home.
One day while at home his spouse finds that it appears he is sleeping at an odd time of day and is rather unarousable. Knowing that he is diabetic she checks his blood sugar using that same glucometer kit. Now this is a broad made up but plausible scenario. Is his spouse an “intended user”? Sure, Matt the diabetic is still a member of the “intended patient population”, but ask yourself some of these follow up questions:
Did the manufacturer of the glucometer kit design and document the intended user to include caretakers of the “intended patient population”?
If not, does this mean that Matt’s spouse was using the glucometer in an off-label manner?
If both caretakers and patients are intended users, are the Instructions For Use written in such a manner that they are clearly understood when applied to testing blood glucose levels on others as well as yourself?
Perhaps this was an unforeseen human factor when designing the glucometer kit that needs further study?
I promise that questions like these are better asked and incorporated into the design and development of a medical device early on rather than having to address them post-market release and have to consider recalls, notifications, corrective actions, etc. in the future.
Do the questions end with my MDR gap analysis?
All of the above discussion resulted from a single sentence, being tweaked just a little bit, in order to make a procedure more clear and leaving less room for interpretation.These are just theoretical questions that should be asked. As the ‘rabbit hole’ always seems to go deeper and branch off so do some of these theoretical situations. This was just a bit of a back and forth conversation with myself regarding a very specific section of Annex II. As we delve deeper into the proverbial rabbit hole, consider again the situation where Matt’s spouse used the device. If she was not an “intended user,” does this qualify as “misuse of the device”? Maybe, or maybe not, but each situation will result in different answers to these questions.
If you go back to Annex I, Chapter 1, Section 3(c) it states, “estimate and evaluate the risks associated with, and occurring during, the intended use and during reasonably foreseeable misuse.” If that is considered misuse, is it ‘reasonably foreseeable’ (taken from the English Version of Regulation EU 2017/745 on 08/31/2018)? What is considered misuse? The EU MDR does not have misuse in its definitions. In fact, the term misuse is only even used three times. To narrow down whether or not this is reasonably foreseeable misuse we need to find a working definition within an accepted harmonized standard or other regulation that applies to the governance of medical devices within the same manner that the EU MDR does.
That same thoroughness needs to be applied to how misuse may be considered foreseeable. Maybe through human factors studies? Maybe through post market surveillance it is discovered that the device is sometimes used by someone other than an intended user, or for something other than the intended purpose. Should misuse be discovered, or suspected does it fall under the realm of it being ‘reasonably foreseeable?’ Ask these questions early, ask them often and then don’t be afraid to ask if they still apply in the future. Have regulations or standards changed? Proactive measures can help discover issues sooner. This lets risks be addressed sooner and ultimately could prevent negative outcomes and experiences from the patients these devices are meant to help.
Conclusions of this MDR gap analysis
I had these thoughts while updating Medical Device Academy’s procedures. First, procedures should always be living documents that can grow and change as standards and regulations metamorphasize to meet the needs of the ever evolving medical device community. This MDR gap analysis applies largely to technical documentation and as such we updated our technical documentation procedure. Every time we analyze quality system documents and technical documentation through the lens of a new standard or regulation, we are certain to expand our appreciation for the complexity of medical device design and development.
What can you do to stay ahead of medical device supply chain disruptions and comply with reporting requirements of possible device shortages?
Supply chain issues can be somewhat cyclical. As we approach the holiday season, we also approach the shipping season. Public shipping services such as FedEx and UPS see an increase in freight as the holiday seasons approach. Manufacturers need raw materials and components to stock the shelves with all of those holiday gifts. Since we are still living under pandemic conditions, I would be willing to bet there will be more care packages and mailed gifts in place of traditional gatherings. On top of the approaching increase in demand, staffing shortages can very quickly exacerbate supply chain bottlenecks. All the while importers are still expected to… well, import! If transportation affects all general industry you can bet it can also cause medical device supply chain disruptions.
So what does an overburdened mail service have to do with medical devices and quality systems?
Consider, how are your customers getting your product in their hands? How are you receiving raw materials and components? How about your contract manufacturer? Do they have supply chain redundancies? Does your supplier quality agreement address notifications for shipping disruptions?
Do you have a regulatory obligation to report a shortage/supply chain disruption or interruption of manufacturing to the FDA, or Health Canada? The FDA monitors for discontinuance and meaningful disruption of manufacturing certain devices and similarly Health Canada monitors their own list of devices for market shortages. Supply chain disruptions either through difficulty sourcing of raw materials and components, or through transportation breakdown of finished devices to market are just one way you could experience a reportable disruption or shortage.
Matthew did not choose the topic of medical device supply chain disruptions randomly. His signature brand of pessimistic cynicism is the reason we have him tasked with keeping his fingers on the pulse of global concerns and potential threats and risks. Potential supply chain disruptions will involve your quality staff in developing preventive actions and contingency plans in case there is an issue. Then, your regulatory team will be in charge of reporting and AHJ notification if you are an affected manufacturer (or importer in Canada!). Understaffed and overloaded shipping and transportation suppliers are about to be bombarded with seasonal freight. This makes them an attractive target for ransomware because, just like healthcare facilities, they will not be in a situation where they can afford any downtime.
Regulatory responsibilities related to device supply chain disruptions – Organized by Jurisdiction
U.S. FDA
The FDA requires reporting shortages and supply chain disruptions to CDHR of permanent discontinuance or interruption in manufacturing of a medical device in Section 506J of the FD&C Act. Especially so in response to the COVID-19 public health emergency. In part, the general public’s need for healthcare during the pandemic guides what devices the FDA needs notification about.
Currently, the FDA is concerned about specific device types by product code or any devices that are critical to public health during a public health emergency. For the most up to date list, the URL to the FDA website will show the specific product codes of the monitored device types;
Health Canada
As an Authority Having Jurisdiction, Health Canada also has reporting requirements for supply chain disruptions of specific types of medical devices. Health Canada is also an independent authority that uses a different device classification system than the U.S. FDA.
The table below shows the device types by their classification level that HC requires supply chain disruption notifications for. This information is current as of September 5th, 2021, and the following link will take you to the HC webpage for the most up-to-date list.
Class I Medical Devices
Masks (surgical, procedure or medical masks) – Level 1, 2, 3 (ATSM)
N95 respirators for medical use
KN95 respirators for medical use
Face shields
Gowns (isolation or surgical gowns) – Level 2, 3 and 4
Gowns (chemotherapy gowns)
Class II Medical Devices
Ventilators (including bi-level positive airway pressure or BiPAP machines, and continuous positive airway pressure or CPAP machines)
Infrared thermometers
Digital thermometers
Oxygen Concentrators
Pulse Oximeters (single measurement)
Aspirators/suction pumps (portable and stationary)
Laryngoscopes
Endotracheal tubes
Manual resuscitation bags (individually or part of a kit)
Medical Gloves – Examination and Surgical (Nitrile, Vinyl)
Oxygen Delivery Devices
Class III Medical Devices
Ventilators (including bi-level positive airway pressure or BiPAP machines)
Harden your supply chain with redundancies. Now is the time to qualify a second supplier as a contingency plan before it is too late…. Maybe even consider opening a Preventive Action? (HINT HINT for those ISO 13485 manufacturers that need to beef up their Clause 8.5.3. operations!)
Supply chains have both up and downstream functions. First, you likely need to source raw materials and components for production. Then you also need to ship those finished devices to distribution centers and your customers. Disrupt either of those and your ability to sell your devices is compromised or even completely halted.
Ask yourself, “Do I have a backup option for shipping?”, and “Do I have a backup option for raw materials and components?”.
Why?
Why go through all of that effort? Well, if you lose UPS and have to use FedEx instead, are their shipping procedures identical? Likely you will need a WI level document for each shipper to explain the process. It is easier to pre-qualify a contingency supplier and establish a WI now rather than in December when holiday shipping is at its peak. Consider if you also need to open accounts, etc. Scheduling pickup online may not be intuitive.
Just identifying a backup is important, but you can take that a step further and pre-qualify them. If they are a shipping and transportation supplier then give them a shipment or two in order to evaluate them. Hold them to the same standards you would for your primary supplier.
Did your shipment arrive on time? Was it damaged during transit? This is provisional, or pre-qualification. Did they perform adequately enough to use as a tentative supplier in the event the primary supplier is unable to perform? This is designed to make a full qualification of this supplier simple and easy… If you need to utilize them that is. Maintaining this pre-qualification should also be simple and easy as well. Once a year or so have them deliver a shipment for you.
That is just for importing or shipping finished devices. Do you have backup raw material or components suppliers identified? If not identifying or even pre-qualifying secondary suppliers might not be a bad idea either. You are probably tied down to a specific geographic area for shipping and transportation. You may not be for raw materials. If you need barrels of silicone consider a backup supplier from a different area than your primary supplier. Natural disasters create havoc for shipping. If your silicone comes from Company A, and they are closed down because of a hurricane then Company B ten miles away is likely affected as well.
For example, if you are in the U.S. and your primary supplier is in the Northeast then a backup supplier in the Southeast may be strategically important. Whereas a backup supplier from the Southwest may be cost-prohibitive.
What about your suppliers? Is your device high-risk enough that if your supply chain is disrupted, you have an obligation to report it to the FDA? In that scenario, if you use a contract manufacturer, it may be worth requiring supply chain contingencies and clearly identifying who owns what reporting responsibilities within your quality agreement with them.
There is an element of proactive responsibility in reporting these shortages, or projected shortages. In order to be able to predict medical device supply chain disruptions, there should be metrics that your quality system is monitoring. What is your monthly production capacity? How much raw material or components does your warehousing have on hand? How many units could you manufacture if the transport industry stopped right this second?
Determine what you need to track in order to identify a disruption before it occurs.
Prepare for notification now. This article looked at the problem from the point of view that transportation issues were the root cause of the supply chain disruption. However, many other things could be disruptive, such as natural disasters and supply availability. Therefore, develop a WI level document for conducting these types of regulatory reporting activities and train personnel before a disruption happens. It is easier to tackle these kinds of problems if you already have process controls in place and trained competent staff than if you wait until the reporting timeline clock is already ticking.
Future blogs about device supply chain disruptions…Shortage Reporting
About the Author
Matthew Walker – QMS, Risk Management, Usability | Human Factors Engineering, Cybersecurity & DFIR
Matthew brings a unique background as a former Firefighter/EMT and Rope Rescue Tech with experience in OSHA and NFPA regulations. For the better part of a decade, he has worked as a Technical/Medical Writer and Lead Auditor. He holds degrees in Fire Science and Computer Forensics and Digital Investigations, graduating Summa Cum Laude from Champlain College. Matthew is also an active member of several academic honor societies including Omicron Sigma Sigma’s Order of the Sword and Shield. His professional focus includes Human Factors Engineering, Risk Management, and Cybersecurity with a special interest in applying Digital Forensics and Incident Response (DFIR) practices to medical technology. He combines regulatory expertise with technical insige to strengthen both product safety and oranizational resiliance. He can be reached by email. You can also follow him on LinkedIn or YouTube.
What is your company’s approach to qualifying a software service provider and managing software-as-a-service (SaaS) for cybersecurity?
The need for qualifying and managing your software service provider
Most of the productivity gains of the past decade are related to the integration of software tools into our business processes. In the past, software licenses were a small part of corporate budgets, and the most critical software tools helped to manage material requirements planning (MRP) functions and customer relationship management (CRM). Today, there are software applications to automate every business process. Failure of a single software service provider, also known as “Software-as-a-Service” or (Saas), can paralyze your entire business. In the past, business continuity plans focused on labor, power, inventory, records, and logistics. Today our business continuity plans also need to expand for the inclusion of software service providers, internet bandwidth, websites, email, and cybersecurity. This new paradigm is not specific to the medical device industry. The medical device industry has become more dependent upon its supply chain due to the ubiquity of outsourcing, and what happens to other industries will eventually filter its way into this little collective niche we share. With that in mind, how do we qualify and manage a software service provider?
Threats to software service providers (Kaseya Case Study)
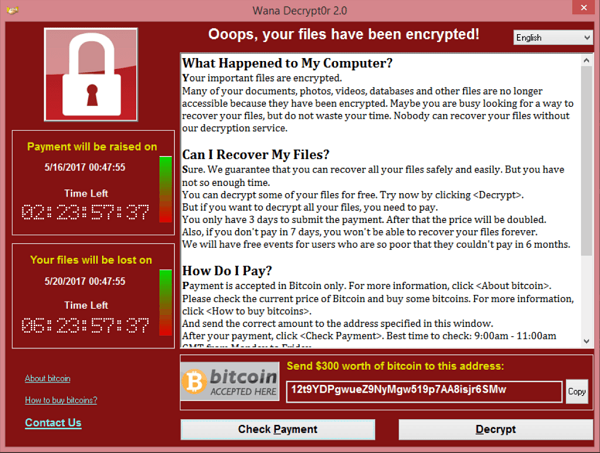
Two years ago the WannaCry ransomware attack affected 200,000 computers, 150 countries, and more than 80 hospitals.
Kaseya isn’t a hospital. Kaseya is a software service provider company. So why is this example relevant to the medical device industry?
The ransomware attack on Kaseya was severe enough that both CISA and the FBI got involved, and it compromised some Managed Service Providers (MSPs) and downstream customers. This supply chain ransomware attack even has its own Wikipedia page. The attack prompted Kaseya to shut down servers temporarily. None of this is a critique of Kaseya or their actions. They were merely the latest high-profile victim of a cyberattack in the news. Now cybercriminals are attacking your supply chain. We want to emphasize the concepts and considerations of this type of attack as it pertains to your business.
What supplier controls do you require for a software service provider?
If you are a manufacturer selling a medical device under the jurisdiction of the U.S. FDA, you need to comply with 21 CFR 820.50 (i.e. purchasing controls). The FDA requires an established and maintained procedure to control how you are ensuring what your company buys meets the specified requirements of what you need. Many device manufacturers only consider suppliers that are making physical components, but a software service provider may be critical to your device if your device is software as a medical device (SaMD), includes software, or interacts with a software accessory. A software service provider may also be involved with quality system software, clinical data management, or your medical device files. Do you purchase software-as-a-service or rely upon an MSP for cloud storage?
You need to determine if your software service provider is involved in document review or approval, controlling quality records, Protected Health Information (PHI), or electronic signature requirements. You don’t need a supplier quality agreement for all of the off-the-shelf items your company purchases. For example, it would be silly to have Sharpie sign a supplier quality agreement because you occasionally purchase a package of highlighters. On the other hand, if you are relying upon Docusign to manage 100% of your signed quality records, you need to know when Docusign updates its software or has a security breach. You should also be validating Docusign as a software tool, and there should be a backup of your information.
21 CFR 820.50 requires that you document supplier evaluations to meet specified and quality requirements per your “established and maintained” procedure. The specified requirements for this supplier might include the following:
How much data storage do you need?
How many user accounts do you need?
Do you need unique electronic IDs for each user?
Do you need tech support for the software service?
Is the software accessed with an internet browser, is the software application-based, or both?
How much does this software service cost?
Is the license a one-time purchase? Or is it a subscription?
The quality requirements for a supplier like this may look more like these questions;
How is my information backed up?
Can I restore previous file revisions in the case of corruption?
How can I control access to my information?
Can I sign electronic documents? If yes, is it 21 CFR Part 11 compliant?
Does this supplier have downstream access to my information? (can the supplier’s suppliers see my stuff?)
Do I manage PHI? If so, can this system be made HIPAA compliant? What about HITECH?
What cybersecurity practices does this supplier utilize?
How are routine patches and updates communicated to me?
A risk-based approach to supplier quality management
ISO 13485:2016 requires that you apply a risk-based approach to all processes, including supplier quality management. A risk-based approach should be applied to suppliers providing both goods and services. For example, you may order shipping boxes and contract sterilization services. Both companies are suppliers, but in this example, the services provided by the contract sterilizer are associated with a much higher risk than the shipping box supplier. Therefore, it makes sense that you would need to exercise greater control over the sterilizer. Software service providers are much like contract sterilizers. SaaS is not tangible but the service provided may have a high level of risk and potential impact on your quality management system. Therefore, you need to determine the risk associated with SaaS before you can evaluate, control, and monitor a software service supplier.
First, you need to document the qualification of a new supplier. It would be nice if your cloud service provider had a valid ISO 13485:2016 certification. You would then have an objectively demonstratable record of their process controls and know that they are routinely audited to maintain that certification. They would also understand and expect to undergo 2nd party supplier audits because they operate in the medical device industry. Alternatively, a software service provider may have an ISO 9001:2015 certification. This is a general quality system certification that may be applied to all products or services. In the absence of quality system certification, you can audit a potential supplier. For some suppliers, this makes sense. However, many companies that are outside of the medical device industry do not even have a quality system because it is not required or typical of their industry. For the ones that do, though, you can likely leverage their existing certifications and accreditations.
Cybersecurity standards you should know
Most cloud service providers will not have ISO 13485 certification, because it is a quality management standard specific to the medical device industry. However, you might look for some combination of the following ISO standards that may be relevant to a software service provider:
ISO/IEC 27001 Information Technology – Security Techniques – Information Security Management Systems – Requirements
ISO/IEC 27002:2013 Information Technology. Security Techniques. Code Of Practice For Information Security Controls
ISO/IEC 27017:2015 Information Technology. Security Techniques. Code Of Practice For Information Security Controls Based On ISO/IEC 27002 For Cloud Services
ISO/IEC 27018:2019 Information Technology – Security Techniques – Code Of Practice For Protection Of Personally Identifiable Information (PII) In Public Clouds Acting As PII Processors
ISO 22301:2019 Security And Resilience – Business Continuity Management Systems – Requirements
ISO/IEC 27701:2019 Security Techniques. Extension to ISO/IEC 27001 and ISO/IEC 27002 For Privacy Information Management. Requirements And Guidelines
Does your software service provider have SOC reports?
The acronym “SOC” stands for Service Organization Control, and these reports were established by the American Institute of Certified Public Accountants. SOC reports are internal controls that an organization utilizes and each report is for a specific subject. SOC reports apply to varying degrees for SaaS and MSP Suppliers
The SOC 1 Report focuses on Internal Controls over Financial Reporting. Depending on what information you need to store on the cloud, this report could be more applicable to the continuity of your overall business than specifically to your quality management system.
The SOC 2 Report addresses what level of control an organization places on the five Trust Service Criteria: 1) Security, 2) Availability, 3) Processing Integrity, 4) Confidentiality, and 5) Privacy. As a medical device manufacturer, these areas would touch on control of documents, control of records, and process validation, among other areas of your quality system. Some suppliers may not share a SOC 2 report with you, because of the amount of confidential detail provided in the report.
The SOC 3 Report will contain much of the same information that the SOC 2 Report contains. They both address the five Trust Service Criteria. The difference is the intended audiences of the reports. The SOC 3 is a general use report expected to be shared with others or publicly available. Therefore, it doesn’t go into the same intimate level of detail as the SOC 2 report. Specifically, information regarding what controls a system utilizes is very brief if identified at all compared to the description and itemized list of controls in the SOC 2 Report.
Other ways to qualify and manage your software service provider
SOC reports will help paint a picture of the organization you are trying to qualify for. You will also need to evaluate the supplier on an ongoing basis. It is essential to know if the supplier is subject to routine audits and inspections to maintain applicable certifications and accreditations. For example, if their ISO certificate lasts for three years, you should know that you should follow up with your supplier for their new certificate at least every three years. On the other hand, if they lose certification, it may signify that the supplier can’t meet your needs any longer and you should find a new supplier.
There is a long list of standards, certifications, accreditations, attestations, and registries that you can use to help qualify a SaaS or MSP supplier. One such registry is maintained by Cloud Security Alliance (i.e. the CSA STAR registry). “STAR” is an acronym standing for Security, Trust, Assurance, and Risk. CSA describes the STAR registry in their own words:
“STAR encompasses the key principles of transparency, rigorous auditing, and harmonization of standards outlined in the Cloud Controls Matrix (CCM) and CAIQ. Publishing to the registry allows organizations to show current and potential customers their security and compliance posture, including the regulations, standards, and frameworks they adhere to. It ultimately reduces complexity and helps alleviate the need to fill out multiple customer questionnaires.”
Some of the questions your supplier qualification process should be asking about your SaaS and MSP suppliers include:
Why do I need this software service?
Which standards, regulations, or process controls need to be met?
What is required for qualifying suppliers providing SaaS or an MSP?
How will you monitor a software service provider?
ISO certification, SOC reports, and the CSA STAR registry are supplier evaluation tools you can use for supplier qualification and monitoring. When you use these tools, make sure that you ask open-ended questions instead of close-ended questions. Our webinar on supplier qualification provides several examples of how to convert your “antique” yes/no questions into value-added questions.
Your software service provider should be able to provide records and metrics demonstrating the effectiveness of their cybersecurity plans. Below are three examples of other types of records you might request:
Cloud Computing Compliance Controls Catalogue or “C5 Attestation Report”
System Security Plan for Controlled Unclassified Information in accordance with NIST publication SP 800-171
Privacy Shield Certification to EU-U.S. Privacy Shield or Swiss-U.S. Privacy Shield
The privacy shield certification may be especially important for companies with CE Marked devices in order to comply with the European Union’s General Data Protection Regulation (GDPR) orRegulation 2016/679.
A final consideration for supplier qualification is, “Who are the upstream suppliers?” It is essential to know if your new supplier or their suppliers will have access to Protected Health Information (PHI). Since you have less control of your supplier’s subcontractors, you may need to evaluate how your supplier manages their supply chain and which general cybersecurity practices your supplier’s subcontractors adhere to.
Additional cybersecurity, software validation, and supplier quality resources
For more resources on cybersecurity, software validation, and supplier quality management please check out the following resources:
Did you know you can download competitor inspectional observations to learn which quality issues are likely to result in warning letters?
Not long ago the FDA published their Inspectional Observation Data Sets. They are Excel spreadsheets of the dreaded 483 inspection observations and warning letters that the FDA issues after performing inspection of manufacturers. There is a spreadsheet for each of the following topic areas, and we will take a look at the ‘Devices’ observations. A post-mortem data analysis or speculative data autopsy if you will… What can we learn when examining an FDA inspection observation?
Biologics
Drugs
Devices
Human Tissue for Transplantation
Radiological Health
Parts 1240 and 1250
Foods (includes Dietary Supplements)
Veterinary Medicine
Bioresearch Monitoring
Special Requirements
Total number of inspections and 483s
These are nonconformities written by the FDA to the Code of Federal Regulations, so there won’t be any statistics for ISO 13485:2016 or Regulation (EU) 2017/745. There will be lots of findings under the ‘QSR’ or 21 CFR 820. The good news, unlike an ISO Standard, is that the Code of Federal Regulations is publicly available online for free. It isn’t a pay-to-play game and we can share the full text of the requirement without violating any copyright licensing agreements.
The top 10 areas for inspection observations and warning letters are:
CAPA procedures
Complaint procedures
Medical Device Reporting
Purchasing Controls
Nonconforming Product
Process Validation
Quality Audits
Documentation of CAPA actions and results
Training
Device Master Record
Corrective and preventive action is the most common reason for warning letters
The winning quality system requirement that resulted in the most 483 inspection observations and warning letters was for Corrective and Preventive Actions under 21 CFR 820.100(a). This finding is listed when a manufacturer fails to establish a CAPA procedure or the procedure is inadequate. This finding was cited 165 times. In addition, CAPA activities or their results were not documented or were not documented adequately a total of 32 times under 21 CFR 820.100(b). This gives us a grand total of 197 observations for the CAPA process.
Corrective and preventive actions are either fixing an identified problem and making sure it doesn’t happen again, or stopping a potential problem from happening in the first place. It is both the reactive and proactive response for quality issues and product non-conformance. The text of the requirement is:
Ҥ820.100 Corrective and preventive action.
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.“
We can see that under section (a) the requirement is that there is an established and maintained process control with a numerical list of required inputs and outputs of that process. The process control is easy, use a procedure. You have to establish a procedure and you have to maintain it. That is one part of the first 165 observations.
The second part is that the procedure needs to be ‘adequate’. That means that bullets (1)-(7) need to be addressed within that procedure. For example number (2) is “Investigating the cause of nonconformities relating to product, processes, and the quality system;”. This means that the procedure should be explaining not only that your quality system will be doing that investigation, but who will be doing it and how they will be doing it.
“The cause of nonconformities shall be investigated”, may not be an adequate process control. Yes, it addressed the need for a root cause evaluation, but does it do that adequately?
“The RA/QA Manager will complete or assign a staff member to complete the root cause evaluation of Corrective Actions utilizing methods such as a ‘5-Why Analysis’ by filling in section 2. Of the CAPA report form.” This wording is much closer to what is needed in a procedure. It explains who is doing what, roughly how they might do it, where that activity gets documented and identifies the record that the activity produces.
Which brings us to the extra 32 findings where the activities and their results either weren’t documented or were done so poorly. This is why identifying the input (Root Cause Analysis) and the output (Section 2. of the CAPA report) are important. It allows you, the inspector or an auditor to trace from the procedure to the record that part of the process produces to demonstrate conformity.
As the age old saying goes, “if it isn’t documented, it didn’t happen”. That record should show that yes you did a root cause analysis (the activity) and what the conclusion of that analysis was (the results of that activity). These types of records are so vital to your quality system that there is an entire process dedicated to the control of records. I’ll give you a hint, it is Subpart-M of the QSR.
This is also a great segway to show how the processes go hand in hand and CAPA is interrelated to Document Control, Record Control, and your Quality System Record. Your system processes will continually wrap back around to each other in this manner. For example, CAPAs are a required input into your Management Review process so if you don’t have a CAPA procedure you aren’t performing adequate management reviews.
A note on other systems
If your quality system is also ISO 13485:2016 compliant, Corrective Actions and Preventive Actions are separate items under separate sub-clauses. Corrective Actions are in 8.5.2., and Preventive Actions are in 8.5.3. Meaning if you have a mature quality system that has never had a preventive action, then your CA might be fine, but the PA of that process may be inadequate.
An industry standard for CAPAs is applying a risk based approach, and we have an entire webinar dedicated to the subject! How to create a risk-based CAPA process
Complaints are the second most common reason for warning letters
The silver medal goes to complaints. Much like CAPA the biggest issue is no, or inadequate complaint handling procedures. This specific finding was cited 139 times (overall complaint handling has more but this specific issue was the most cited). Not to sound like a broken record but again, complaint handling is a specific process that requires an ‘established and maintained procedure”.
As a procedure it has to exist, it has to be maintained, and each process has requirements for inputs and outputs that must be outlined. Complaint handling is a little bit different in the QSR in that there isn’t a ‘complaint’ sub-part. Complaints are under Sub-Part M- Records, specifically 21 CFR 820.198 Complaint Files.
To compare, Complaints in accordance with ISO 13485:2016 are under Measurement Analysis and Improvement, specifically Sub-clause 8.2.2. Complaint Handling. It is sandwiched in between Feedback and Reporting to Regulatory Authorities. That had to have been done on purpose because those processes are inherently intertwined and their inputs and outputs directly feed into each other:
Ҥ820.198 Complaint files.
(a) Each manufacturer shall maintain complaint files. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit. Such procedures shall ensure that:
(1) All complaints are processed in a uniform and timely manner;
(2) Oral complaints are documented upon receipt; and
(3) Complaints are evaluated to determine whether the complaint represents an event which is required to be reported to FDA under part 803 of this chapter, Medical Device Reporting.”
This sub-section of ‘Records’ may be less intuitive than what we saw under CA/PA. We can see that we have to maintain complaint files. We also need a procedure that covers receipt, review, and evaluation of complaints. Then we have to name a formally designated complaint handling unit to do all of that.
Further we need to make sure that complaints are handled uniformly and efficiently. It should be a cookie cutter process with a known timeline. Every complaint goes through the same review and evaluation within a specific time period. If it takes six months to review a complaint, that definitely is not a ‘timely manner’.
Not every complaint will be sent to you via certified mail with ‘Complaint’ written across the top in big BOLD letters. Sometimes people will simply tell you about a complaint they have verbally and your process needs to define how it is addressing these verbal communications. Otherwise your FDA inspection observation will be written, and you run the risk of receiving warning letters.
This of course begs the question, what is a complaint? How will I know if I received one? Fortunately 21 CFR 820.3 provides us with definitions, one of them being what exactly a complaint is “(b) Complaint means any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution.”.
There is no quiz at the end of this but I would caution you that this will probably be on the test. Anytime you ask a question like that and the regulation provides a definition for it, then it’s a good idea to include that definition within your procedure. This is a way to make sure that there is uniformity in the understanding of a procedure. If you miss a complaint because you didn’t realize that it was a complaint then your process is not effective. Eventually an auditor will pick up on the deficiencies in the process, document a finding and you will be doing a CAPA to fix it.
Every complaint needs to be reviewed, but not every complaint needs to be investigated. This was a much less cited issue (5). You are allowed to decide that an investigation isn’t needed. However, if you do then you must keep a record of why you decided that and name the person responsible for that decision.
That isn’t carte’ blanche to just write off investigations whenever you want. There are some things that require an investigation and there is no accepted rationale for not performing one. An example is when there is a possible failure of a device, it’s labeling or packaging to meet any of their specifications. Those need to be investigated without exception. What your system is allowed to do though is if you have already done an investigation and you received related similar complaints, there is no need to repeat the same investigation for every complaint.
An important concept of complaint handling is that you should be triaging your complaints as you receive them. There are certain types of complaints that must be reported to the FDA. More information is actually found under 21 CFR 803, not the 820 that we have been examining. These special complaints need to be identifiably separate from your normal run of the mill complaints. These complaints specifically need a determination of;
Whether the device failed to meet specifications;
Whether the device was being used for treatment or diagnosis; and
The relationship, if any, of the device to the reported incident or adverse event.
Outside of those special reportable complaints, all investigations have certain required outputs. By addressing every complaint in a uniform repeatable manner, this can be boiled down to a form. In fact creating a specific complaint form makes sure that all of the required information has been documented. Each record of an investigation by your formally designated complaint handling unit has to be include;
The name of the device;
The date the complaint was received;
Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used;
The name, address, and phone number of the complainant;
The nature and details of the complaint;
The dates and results of the investigation;
Any corrective action taken; and
Any reply to the complainant.
Some companies and corporations sprawl across the globe and have many sites all over the place. Not every manufacturer is limited to containing all of their operations within a single building. There are times where the formally designated complaint handling unit may be somewhere other than where the manufacturing is taking place. That is acceptable as long as communication between the two is reasonably acceptable. The manufacturer needs access to the records of the complaint investigations performed. Just as everything must be documented, all of that documentation must be producible as well. If not, your inspector will produce FDA 483 inpsection observations and warning letters.
If the complaint handling unit is outside of the United States the records have to be accessible in the United states from either the place where the manufacturers records are normally kept or at the initial distributor.
Complaint Handling and vigilance reporting are topics that we often find stuck together like velcro. We find them so interelated that we have a combined Complaint Handling and Vigilance Reporting Webinar.
Medical Device Reporting is the third most common reason for warning letters
The bronze medal recipient shows a drop in sheer numbers of FDA inspection observations. A total of 68 were written for the fiscal year of 2020, and these findings have a high likelihood of resulting in warning letters because these incidents may involve serious injuries and death. We are slowing down, but this is still a topic that gets an FDA inspection observation almost every week.
But again part of the issue is no, or bad procedures to control this process. Not to be confused with the (EU) MDR since as an industry we love acronyms so much, Medical Device Reporting is referenced within the Quality System Requirements of 21 CFR 820. We took a peek above in Complaint Handling. What makes this unique is that MDR actually lives in 21 CFR 803 Medical Device Reporting. What makes it even more special is that Part 803 is further broken down into sub-parts.
We will take a look at Sub-part E which is the reporting requirements for manufacturers. Medical Device Reporting is a process and as such needs a procedure to control it and that procedure must be maintained.
Some key points to capture is that there are reporting timelines that are measured in calendar days from when you become aware of information that reasonably suggests that one of your devices;
“(1) May have caused or contributed to a death or serious injury or (2) Has malfunctioned and this device or a similar device that you market would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur.”
There are some crucial takeaways. First, the clock starts ticking down calendar days, not work days, and holidays count. You can’t hold off reporting that your device killed someone because it’s around the holidays and over a few weekends.
Second, is that reporting timelines vary, generally between 5 and 30 calendar days. That means it is important to know the specific timeline for the type of report you are making and what the authority having jurisdiction requires for a timeline. The FDA may differ from Health Canada which in turn may differ from the EU, etc.
Third is that the bar to meet is what would be ‘reasonably known’, and that is somewhat of an ambiguous requirement open to interpretation.
They help clarify this with,
“(i) Any information that you can obtain by contacting a user facility, importer, or other initial reporter; (ii) Any information in your possession; or (iii) Any information that you can obtain by analysis, testing, or other evaluation of the device.”
The first two are usually not an issue, but the one that tends to get less attention is deeper analysis, testing or evaluation of the device. Due diligence is required here to make sure that you actually do know the information that should be ‘reasonably known’ to you.
The burden of investigation and root cause determination is placed squarely on the shoulders of the manufacturers and that is a process that can take some time. What happens when the reporting timely is fast approaching but your investigation won’t be finished before the clock runs out? The short answer is to report it anyway.
The longer answer is to report what information you do have with an explanation of why the report doesn’t have all of the required information. Then explain what you did to try to get all of the information, and file a supplemental or follow-up report later to fill in the gaps. Only having a partial report ready is not an excuse to miss the reporting deadline. It is however, the perfect excuse to get an FDA inpsection observation or warning letters.
This article helps you understand how to pass the FDA Refusal to Accept (RTA) screening process 510k submissions – updated April 2022.
What is an RTA Checklist?
The “RTA” in RTA Checklist stands for Refuse to Accept. The FDA uses this tool to determine if your 510(k) submissions will be accepted or not for a substantive review. Accepted, not approved because this is simply a verification that the required information is included in your submission. As stated in the 2022 FDA guidance document for the FDA’s Refuse to Accept Policy for 510(k)s “a minimum threshold of acceptability and should be accepted for substantive review.”(Ref.1). That does a nice job summarizing the RTA checklist. It is a tool used to help assess whether or not your submission contains the required information to continue with a more thorough review of the contents of the submission itself.
What does the Refusal to Accept (RTA) policy apply to?
The Refusal to Accept (RTA) policy applies to all 510k submissions. The RTA checklist or more checklists apply specifically to each 510(k) submission type:
Traditional 510k
Abbreviated 510k
Special 510k
There is a different RTA checklist for each submission type. The checklists are in the Refuse to Accept Policy for 510(k)s guidance document. Specifically, in the PDF document that the FDA reissued in April 2022, the checklists can be found in the following areas:
Traditional 510k – Appendix A.
Abbreviated 510k – Appendix B.
Special 510k – Appendix C.
Note that the title of the checklist refers to an ‘acceptance checklist.’ It is not called the RTA checklist until you get to the footer of the page. It is also listed as an acceptance checklist on the FDA website. The best way to think of the process is as preliminary screening by the FDA.
What does the FDA look at during the Refusal to Accept (RTA) screening process?
During the screening process, the assigned RTA screener will review the 510k submission and try to identify all of the requirements listed in the applicable RTA checklist. The person screening your submission is required to answer “yes,” “no,” or “n/a” to the questions in the checklist. This person must also enter the document and the page where the information can be found in the submission. Finally, if an element required by the refusal to accept (RTA) checklist cannot be found, then the screener adds a comment at the end of that section in the checklist. The comment will state what your deficiency is and it may even identify a guidance document that can help you address the issue. If you are missing requirements, you will receive an email from the RTA screener with the completed RTA checklist attached. We call this an “RTA Hold” letter. If your submission is not rejected, then your 510k is administratively complete, and you will receive an automated email indicating that your submission was accepted and the substantive review will begin.
Refusal to Accept (RTA) Time Frame
As stated in the guidance document, the Refusal to Accept policy includes “an early review against specific acceptance criteria and to inform the submitter within the first 15 calendar days after receipt of the submission if the submission is administratively complete, or if not, to identify the missing element(s).” (Ref. 1). If the assigned screening person is unable to complete the process within 15 calendar days, then you will receive an automated email stating that they were unable to complete the RTA checklist within 15 calendar days, and your submission is automatically moved to the substantive review stage of the 510k review process.
Taking the time to perform your gap analysis before submitting could avoid a simple error. For example, if you forget to include the signed Truthful and Accuracy Statement in your submission, it could take 15 days to be notified of that missing element. The person screening your submission could email you to provide this missing element in an interactive review to avoid placing your submission on hold. Still, they are not required to give you a chance to provide this interactively by email. If you do receive an RTA Hold letter, you might be able to correct missing elements on the same day, but the 510k review clock is automatically reset when your 510k is placed on RTA Hold. When you respond to an RTA Hold letter, there will be another 15-day refusal to accept (RTA) screening of your submission.
What do you do with the information in the comments of the RTA checklist?
The RTA checklist is the criteria that your submission is being evaluated against. Suppose your submission has deficiencies during the initial review against the RTA Checklist. In that case, the FDA will refuse to accept it, and the substantive review will not begin until those deficiencies have been corrected. Since the FDA does not hide what they are looking for or how they will evaluate your submission, use that to your advantage. Assuming that you have correctly determined the type of 510k submission you have, perform a gap analysis of your submission against the RTA checklist. Either perform these actions in-house or hire an outside consultant to do them for you, but make sure you don’t try to check your own work because you will miss something.
Scope of the FDA Refusal to Accept Guidance Document
The scope of the FDA guidance document is provided for the benefit of the FDA personnel reviewing your submission and not specifically for the 510k submitter. It also provides a loose framework for systematically and consistently reviewing submissions. This ensures all submissions receive equal, nonbiased treatment. There are some things that this guidance document does not address or alter by its own admission. One is the “substantial equivalence decision-making process once the submission has been accepted for review.” Refusing to accept (RTA) guidance also does not address FDA user fees. Other guidance documents address those issues.
What are the most common reasons for FDA refusal of your 510k submission?
Although there are dozens of reasons (43 to be exact) why the FDA could reject your submission in the 35-page RTA checklist, most of the refusals (~80%) result from a small percentage (~20%) of reasons. The most common is that your submission is poorly organized. Either you did not provide a table of contents, your submission is not organized in accordance with the sections outlined in the guidance, or the pages of your submission are not properly numbered. When trying to review a 1,200-page submission, poor organization is extremely irritating and wastes the reviewer’s time. If it were my decision, I would refuse to complete the entire checklist until you gave me a properly organized submission.
The second most common reason for refusal is submitting a device description that is inadequate. The FDA needs more detail than most companies provide for the device description because they need to understand the differences between your device and the predicate device. This includes much more than just the indications for use. Who are the intended patients and users? What is the intended environment of use? What are the materials for patient-contacting components? What is the source of power for your device? Which design features does your device include when compared to the predicate? What is the user interface for your device? Which accessory devices are needed with your device? You can even make the mistake of being inconsistent in your submission by not repeating the content in the device description in other sections of the 510k submission. It is important to duplicate certain content verbatim in other documents, such as the 510k summary, the executive summary, the substantial equivalence comparison, and the instructions for use. Paraphrasing and summarizing certain information will not work.
The third most common reason for refusal of your submission is likely related to software validation documentation. In addition to complying with the recognized IEC 62304 standard, you must also comply with the five software guidance documents published by the FDA. The FDA and 3rd-party reviewers use an 11-item checklist based on the 2005 FDA guidance document on software validation documentation. In addition, if your device has any of the following five elements, your submission must also comply with the two FDA guidance documents on cybersecurity:
Cloud communication
Network connection (active or not)
Wireless communication in any form
USB/serial ports/removable media
Software upgrades (this includes patches)
Finally, biocompatibility is the one testing section of your 510k submission that is most likely to result in refusal to accept by the FDA out of the seven sections requiring testing reports. There are several reasons why biocompatibility results in more refusals than the other six testing sections. First, the FDA requirements go above and beyond the ISO 10993-1 standard requirements. Second, the FDA requires that you submit full testing reports for biocompatibility, while you can submit summaries for other sections (e.g., sterilization validation). Third, many submitters try to provide a rationale for why testing is not required for their device. Still, the FDA has very stringent requirements for the use of a biological risk assessment or a biocompatibility certification statement in lieu of testing.
Do you have to follow the RTA checklist exactly?
You can, but you are also not bound by it. Like all guidance documents, they “contain nonbinding recommendations”. The checklist is released as part of a guidance document, so it is a guidance and not a regulatory requirement. That being said, if your submission is missing an element in the checklist, your 510k submission will be considered administratively incomplete unless you provide a clear explanation as to why the checklist element does not apply to your submission or you explain how you meet the 510k submission requirement in another way.
Medical devices vary wildly, and there is no one size fits all approach. The FDA recognizes that and includes some wiggle room that gives them some discretion in reviewing submissions. However, 100% of the 3,500+ submissions received each year are screened using the refusal to accept (RTA) checklist, and the screening person’s job is to verify that your submission meets the criteria. As it says in the guidance document:
“The purpose of the 510(k) acceptance review is to assess whether a submission is administratively complete, in that it includes all of the information necessary for FDA to conduct a substantive review. Therefore, the submission should not be accepted and should receive an RTA designation if one or more of the items noted as RTA items in the checklist are not present and no explanation is provided for the omission(s). However, during the RTA review, FDA staff has the discretion to determine whether missing checklist items are needed to ensure that the submission is administratively complete to allow the submission to be accepted. FDA staff also has the discretion to request missing checklist items interactively from submitters during the RTA review. Interaction during the RTA review is dependent on the FDA staff’s determination that outstanding issues are appropriate for interactive review and that adequate time is available for the submitter to provide supporting information and for FDA staff to assess responses. If one or more items noted as RTA items on the Acceptance Checklist are not present, FDA staff conducting the acceptance review should obtain management concurrence and notify the designated 510(k), contact person, electronically that the submission has not been accepted. “ (Ref. 1).
The portion above notes that explanations may be provided for omitted portions of the submission. So, the answer to the question is that no, you do not have to follow the RTA checklist exactly. However, if you should purposefully omit a section you should provide an explanation and your rationale justifying why the omission is appropriate for your individual device and 510(k) submission. Again, just because you have included an alternative approach or justification does not automatically mean it will be accepted. The FDA personnel who are conducting the acceptance review will judge whether or not your deviation is acceptable.
What if your 510k submission is refused?
If your submission is refused, you will be provided with a copy of the completed RTA checklist, and each of the deficiencies you must address will be highlighted. Sometimes, there will be an attachment to the checklist that has additional issues that are not in the RTA checklist, but the reviewer thinks you may need to address them later. You might also see comments that are not highlighted. These are suggestions from the reviewer that you may or may not choose to address.
There is a 180-day timeline for response to an RTA Hold letter. The response must be submitted to the CDRH Document Control Center (DCC) as an eCopy, and the response must be received within 180 days. If the response is not received within 180 days, your submission will be automatically withdrawn on the 181st day. Your response may not be piecemeal. You must address all of the issues in the RTA checklist or your submission will be placed on RTA Hold again (i.e., RTA2). If you are not sure how to organize your response, a previous blog posting and YouTube video address this topic directly.
About the Author
Matthew Walker – QMS, Risk Management, Usability | Human Factors Engineering, Cybersecurity & DFIR
Matthew brings a unique background as a former Firefighter/EMT and Rope Rescue Tech with experience in OSHA and NFPA regulations. For the better part of a decade, he has worked as a Technical/Medical Writer and Lead Auditor. He holds degrees in Fire Science and Computer Forensics and Digital Investigations, graduating Summa Cum Laude from Champlain College. Matthew is also an active member of several academic honor societies including Omicron Sigma Sigma’s Order of the Sword and Shield. His professional focus includes Human Factors Engineering, Risk Management, and Cybersecurity with a special interest in applying Digital Forensics and Incident Response (DFIR) practices to medical technology. He combines regulatory expertise with technical insige to strengthen both product safety and oranizational resiliance. He can be reached by email. You can also follow him on LinkedIn or YouTube.
The US FDA does not require that 100% of your quality system be implemented before 510k clearance, but these 10 activities need to be done.
The form above allows you to register for a live webinar we are hosting on Friday, May 21, 2021 @ 1 pm EDT. The webinar will share the 510k project management lessons learned by our team since 2016. In addition to 510k project management, MedTech companies also need to implement their quality system in parallel with their regulatory submissions. Some people say that you need to implement your quality system before you submit your 510k. That is not an FDA requirement, but you do have quality system activities that need to be done before you will have all of the technical documentation you need to submit a 510k. This article describes 10 quality tasks you need to prevent unexpected delays.
Design & Risk Management Planning
Design & Risk Management Planning is your 1st priority because you want to identify all of the major activities that need to be completed in your design and risk management processes and which activities are critical path items. Otherwise, you will have unexpected delays. You can and should add details to the plan as you go, but items 2-9 listed below should be included in that initial plan–along with your design and risk management activities.
Risk Management Activities are Needed Before 510k Clearance
Formative Usability Testing is your 3rd priority because this helps you evaluate your device design while it’s still evolving. Formative testing helps you identify opportunities for improvement, provides confirmation that your design is moving in the right direction, and identifies potential use errors while there is still time to implement effective risk controls such as alarms and other safety features. References:
Software Validation is Needed Before 510k Clearance
Software Validation is your 4th priority because it must precede electrical safety testing for electromedical devices and most companies underestimate the time required to document software validation in accordance with IEC 62304:2006 / AMD 1:2015 and the FDA’s five guidance documents:
Supplier Qualification is Needed Before 510k Clearance
Supplier qualification is your 5th priority because you do not want to order all of your prototype parts for the initial testing parts and then find out that the supplier is not capable of supporting you commercially. If you have to switch suppliers you might be forced to repeat biocompatibility testing and other design verification testing due to changes in the manufacturing process. Implementation of a supplier qualification process before 510k clearance is needed.
Label & IFU Requirements Specifications
Label requirements and instructions for use requirements specifications is your 6th priority because you cannot perform electrical safety testing or design validation (including summative usability testing) of your device without labeling and instructions. These requirements are the design inputs for information provided to the user and these must be controlled under design controls rather than document control.
Packaging Specifications
Packaging specifications is the 7th priority you should implement before 510k clearance because the packaging is needed to maintain sterility, to ensure product stability, and to protect the product from shipping. Companies are also frequently surprised by the long lead times associated with ordering custom packaging and you may not have the budget to validate sub-optimal “stock” packaging for your 510(k) submission and then repeat the validation for the optimized packaging later.
Quality System Implementation
Quality system implementation is the 8th priority for implementation before 510k clearance because you will need a fully functional quality system by the time your 510(k) is cleared. Quality system implementation typically takes 6+ months while the 510(k) review should take 4 months or less. Quality system implementation includes writing 25+ procedures, reviewing and approving those procedures, training your employees, and actually using those procedures to begin generating quality system records. For companies that are pursuing Canadian Licensing or CE Marking, the quality system must be fully implemented and certified before the regulatory submission is possible. (Quality System Requirements for the U.S. FDA are outlined within 21 CFR 820-Quality System Regulation)
Apply for Small Business Status Before 510k Clearance
Application for small business status should be the 10th priority for implementation before 510k clearance because this can save your company $16,000+ but it requires that you submit your application at least 60 days before you need to pay the 510(k) user fee.
About the Author
Matthew Walker – QMS, Risk Management, Usability | Human Factors Engineering, Cybersecurity & DFIR
Matthew brings a unique background as a former Firefighter/EMT and Rope Rescue Tech with experience in OSHA and NFPA regulations. For the better part of a decade, he has worked as a Technical/Medical Writer and Lead Auditor. He holds degrees in Fire Science and Computer Forensics and Digital Investigations, graduating Summa Cum Laude from Champlain College. Matthew is also an active member of several academic honor societies including Omicron Sigma Sigma’s Order of the Sword and Shield. His professional focus includes Human Factors Engineering, Risk Management, and Cybersecurity with a special interest in applying Digital Forensics and Incident Response (DFIR) practices to medical technology. He combines regulatory expertise with technical insige to strengthen both product safety and oranizational resiliance. He can be reached by email. You can also follow him on LinkedIn or YouTube.
This blog is intended to help clear your justified confusion if you are wondering what the difference is between PMS, PSUR, and PSR.
The nine most terrifying words in the English language are, “I’m from the Government, and I’m here to help.” That quote is from a speech by President Reagan on August 12, 1986. One of the goals of the European Parliament and Council was “to ensure effective coordination of [competent authority] market surveillance activities and to clarify the applicable procedures.” After studying the new European MDR, I can confidently say that the European Parliament and Council have done their job well. My boss is a regulatory consultant with 30 years of experience, and he asked me to explain the difference between PMS, PSUR, and PSR.
To answer that question as objectively as possible, and cite my sources, I have included a copy and paste directly from Regulation (EU) 2017/745. Red text is my commentary, while the italicized text is a quotation from the most relevant article within the new EU regulations.
Under the New MDR, the only Class IIa, Class IIb, and Class III products are definitively required to have a Periodic Safety Update Report (PSUR). The PSUR needs to be updated annually for Class III and Class IIb implants, and the PSUR needs to be updated at least every two years for Class IIb (non-implants) and Class IIa devices. The PSUR must be available to your notified body, and upon request, the competent authorities. In contrast with the PSUR, Post-Market Surveillance (PMS) reports are required for Class I devices. Finally, a manufacturer’s Periodic Summary Report (PSR), relates to specific cases of Serious Incidents and Field Safety Corrective Actions (FSCA’s) based upon an agreement between the manufacturer and the competent authority or authorities instead of submitting individual FSCA reports. This is confusing because the PSUR also meets the requirements of a PMS Report as defined in Article 85, but we don’t call it a PMS Report.
“Article 83 – Post-market surveillance system of the manufacturer
1. For each device, manufacturers shall plan, establish, document, implement, maintain, and update a post-market surveillance system in a manner that is proportionate to the risk class and appropriate for the type of device. That system shall be an integral part of the manufacturer’s quality management system referred to in Article 10(9).”
In Matthew’s words, “Manufacturers are required to establish a PMS system for every device or device family.”
“Article 84 – Post-market surveillance plan
The post-market surveillance system referred to in Article 83 shall be based on a post-market surveillance plan, the requirements for which are set out in Section 1.1 of Annex III. For devices other than custom-made devices, the post-market surveillance plan shall be part of the technical documentation specified in Annex II.”
In Matthew’s words, “Article 84 requires you to have a PMS plan in your quality system.”
“Article 85 – Post-market surveillance report
Manufacturers of class I devices shall prepare a post-market surveillance report summarizing the results and conclusions of the analyses of the post-market surveillance data gathered as a result of the post-market surveillance plan referred to in Article 84 together with a rationale and description of any preventive and corrective actions taken. The report shall be updated when necessary and made available to the competent authority upon request.”
In Matthew’s words, “A Class I device requires a PMS report, while the other product classifications require a PSUR.”
“Article 86 – Periodic safety update report
1.1 – Manufacturers of class IIa, class IIb, and class III devices shall prepare a periodic safety update report (‘PSUR’) for each device and were relevant for each category or group of devices summarizing the results and conclusions of the analyses of the post-market surveillance data gathered as a result of the post-market surveillance plan referred to in Article 84 together with a rationale and description of any preventive and corrective actions taken. Throughout the lifetime of the device concerned, that PSUR shall set out:
(a)
the conclusions of the benefit-risk determination;
(b)
the main findings of the PMCF; and
(c)
the volume of sales of the device and an estimated evaluation of the size and other characteristics of the population using the device and, where practicable, the usage frequency of the device.
Manufacturers of class IIb and class III devices shall update the PSUR at least annually. That PSUR shall, except in the case of custom-made devices, be part of the technical documentation as specified in Annexes II and III.
Manufacturers of class IIa devices shall update the PSUR when necessary and at least every two years. That PSUR shall, except in the case of custom-made devices, be part of the technical documentation as specified in Annexes II and III.
For custom-made devices, the PSUR shall be part of the documentation referred to in Section 2 of Annex XIII.
For class III devices or implantable devices, manufacturers shall submit PSURs by means of the electronic system referred to in Article 92 to the notified body involved in the conformity assessment in accordance with Article 52. The notified body shall review the report and add its evaluation to that electronic system with details of any action taken. Such PSURs and the evaluation by the notified body shall be made available to competent authorities through that electronic system.
For devices other than those referred to in paragraph 2, manufacturers shall make PSURs available to the notified body involved in the conformity assessment and, upon request, to competent authorities.”
In Matthew’s words, “Barring specified exemptions, manufacturers of a Class IIa device would need to submit a PSUR and update it at least every two years.”
“Article 87 – Reporting of serious incidents and field safety corrective actions
9. For similar serious incidents that occur with the same device or device type and for which the root cause has been identified or a field safety corrective action implemented or where the incidents are common and well documented, the manufacturer may provide periodic summary reports instead of individual serious incident reports, on condition that the coordinating competent authority referred to in Article 89(9), in consultation with the competent authorities referred to in point (a) of Article 92(8), has agreed with the manufacturer on the format, content, and frequency of the periodic summary reporting. Where a single competent authority is referred to in points (a) and (b) of Article 92(8), the manufacturer may provide periodic summary reports following an agreement with that competent authority.”
In Matthew’s words, “Periodic summary reports (PSRs) refer to significant incidents (SIs) and field safety corrective actions (FSCAs). PSRs require an agreement between the manufacturer and the competent authority(s) for cases where there is a group of common, well-known, and documented SIs or FSCA’s with a known root-cause. PSRs are an alternative to submitting individual SI and FSCA reports.”
Additional Quality System Resources
My boss also asked me to update the procedures for post-market surveillance (SYS-019) and vigilance (SYS-036). The PMS procedure includes requirements for Articles 83-86. The vigilance procedure includes the requirements for Articles 87-92.
About the author
Matthew Walker – QMS, Risk Management, Usability | Human Factors Engineering, Cybersecurity & DFIR
Matthew brings a unique background as a former Firefighter/EMT and Rope Rescue Tech with experience in OSHA and NFPA regulations. For the better part of a decade, he has worked as a Technical/Medical Writer and Lead Auditor. He holds degrees in Fire Science and Computer Forensics and Digital Investigations, graduating Summa Cum Laude from Champlain College. Matthew is also an active member of several academic honor societies including Omicron Sigma Sigma’s Order of the Sword and Shield. His professional focus includes Human Factors Engineering, Risk Management, and Cybersecurity with a special interest in applying Digital Forensics and Incident Response (DFIR) practices to medical technology. He combines regulatory expertise with technical insige to strengthen both product safety and oranizational resiliance. He can be reached by email. You can also follow him on LinkedIn or YouTube.
This article breaks down and reviews the new implant card requirement as well as Article 18 of EU 2017/745.
We also have available for sale, SYS-037 Implant Card Procedure written to be Article 18 compliant of Regulation (EU) 2017/745, and includes;
SYS-037 A, Implant Card Procedure
FRM-044 Checklist for Information to be supplied to the patient with an implant
FRM-045 Implant Card Checklist for Article 18 Reg 2017-745
Native Slide Deck for Implant Card Webinar
Recording of the Implant Card Webinar
Implant Card Requirement, a new requirement from Regulation (EU) 2017/745.
One of the new changes to the regulation is an introduction of a new requirement for implantable devices. These devices must now come with an “implant card” that contains information about the implanted medical device for the patient. The responsibility of the implementation of the new implant card rules lies with the manufacturer of the implantable device and the health institution as required by the EU member states.
What is an implantable device?
Before discussing the specifics of the implant card, we must first define what an implantable device is to determine if the implant card requirements apply to your device or devices. Article 2 Definitions, number 5 of Regulation (EU) 2017/745 defines and outlines what is considered an implantable device.
“(5) ‘implantable device’ means any device, including those that are partially or wholly absorbed, which is intended:
– to be introduced in the human body, or
– to replace an epithelial surface or the surface of the eye,
By clinical intervention and which is intended to remain in place after the procedure.
Any device intended to be partially introduced into the human body by clinical intervention and intended to remain in place after the procedure for at least 30 days shall also be deemed to be an implantable device; “
Working with the above definition of an implantable device, you can now compare those requirements against your own devices to determine if they are considered to be an implantable device or not. This can be done by performing a gap analysis of the definition against your device.
Consider what your device is and ask yourself the following questions:
Is my device intended to be partially or wholly absorbed?
If the answer is no, then your device may not be an implantable one. If it is, then you must keep asking yourself questions until you can sufficiently determine your device’s status as implantable or not.
Is my device intended to be introduced in the human body?
No. Ok, that is fine, but is it intended to replace an epithelial surface or the surface of the eye?
To make an awful analogy of the process, it is almost like playing a game of Guess Who with your device. Instead of asking your device if they have red hair or a mustache, you have to ask your device questions like, “Are you intended to remain in place after the procedure?”.
The gap analysis is fine, but you also have to consider some other factors within the wording of the definition. Be careful navigating the specifics because the devil is in the details. In the definition, which is only eighty-nine words long, by the way, uses the word “intended” three different times.
That is important because the definition applies not only to some of the characteristics and uses of the device but also to the intent behind the device. Just because the device can be wholly introduced into the body does not mean that the device is ‘intended’ to be. A better example would be, by clinical intervention, can your device remain in place after the procedure? Could it, perhaps, but is it intended to be? Also, is it the intent of the device to be done so by clinical intervention?
Where to find the implant card requirement?
Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices is where the introduction of implant cards can be found. The definition of an implantable device is found in Article 2 Definitions, definition number 5.
Article 18- ‘Implant card and information to be supplied to the patient with an implanted device’ is where the implant card requirements can be found. This article contains three sections and four subsections pertaining to implant cards.
Article 18 Implant card requirement and information to be supplied to the patient with an implanted device
Below is article 18 in its entirety so that we can discuss it further in detail.
“1. The manufacturer of an implantable device shall provide together with the device the following:
(a)
information allowing the identification of the device, including the device name, serial number, lot number, the UDI, the device model, as well as the name, address and the website of the manufacturer;
(b)
any warnings, precautions or measures to be taken by the patient or a healthcare professional with regard to reciprocal interference with reasonably foreseeable external influences, medical examinations or environmental conditions;
(c)
any information about the expected lifetime of the device and any necessary follow-up;
(d)
any other information to ensure the safe use of the device by the patient, including the information in point (u) of Section 23.4 of Annex I.
The information referred to in the first subparagraph shall be provided, to make it available to the particular patient who has been implanted with the device, by any means that allow rapid access to that information and shall be stated in the language(s) determined by the concerned Member State. The information shall be written in a way that is readily understood by a layperson and shall be updated where appropriate. Updates of the information shall be made available to the patient via the website mentioned in point (a) of the first subparagraph.
Also, the manufacturer shall provide the information referred to in point (a) of the first subparagraph on an implant card delivered with the device.
The Member States shall require health institutions to make the information referred to in paragraph 1 available, by any means that allow rapid access to that information, to any patients who have been implanted with the device, together with the implant card, which shall bear their identity.
The following implants shall be exempted from the obligations laid down in this Article: sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors. The Commission is empowered to adopt delegated acts in accordance with Article 115 to amend this list by adding other types of implants to it or by removing implants therefrom.”
Section 1. of Article 18 states explicitly that it is the manufacturer who shall supply the information. Fortunately, it is also outlined what information needs to be included and some guidance on how to provide the information.
Take note, though, that the article states it “shall” be provided, “together with the device.” This means that merely having the information available or accessible such as a downloaded PDF on your website, is not sufficient to comply with section 1. Because that is not being supplied together with the device as outlined.
Section 2. of Article 18 applies to member states’ requirements of health care institutions.
Section 1 of Article 18
Section 1 is by far the most extended section of the article and outlines precisely what information must be provided with the implantable device. Not only is this information that must be provided, it specifically must be provided by the manufacturer. The subsections are broken down by topic and can be summarized as the information, warning, maintenance, and misc. Sections.
Section 1. Sub-Section A
This sub-section outlines the specific identifying information that must be provided. It is even specifically “information allowing the identification of the device.” For devices that are produced and manufactured compliant with other standards such as ISO 13485 or the QSR portion of the United States Code of Federal Regulations, a lot of this information is the same information that is required for traceability.
Besides the generic “information allowing the identification of the device,” the other specific information that ‘shall’ be provided is:
The name of the device,
The device serial number,
The lot number of the device,
The UDI,
The model of the device,
The name of the manufacturer,
The manufacturers address,
The manufacturers’ website.
They don’t just want your device’s driver’s license; they want the driver’s license, library card, passport, blood type, and favorite color. This is done for a purpose but also carries some implications on the maintenance actions of the manufacturer.
First such strict ID requirements mean that the device is traceable and identifiable. There should be absolutely no doubt about who made the device. In the event of an incident, that device should be traceable back to when and where the individual components were created and assembled into the final device. For traceability of an incident, tracking for corrective or preventive action, or just general inventory tracking this is the type of strict diligence that is expected when the end-user or patient is receiving medical care with an implantable device. There is no demonizing of this requirement. Yes, it is strict, but it is also just part of good housekeeping for a manufacturer in general. Only now it must be provided to the patient receiving care with the device as well.
What is implied is that the information provided along with the device is somewhat of a living document, and the information could vary a bit from patient to patient. Because things like lot numbers or any number of trackable metrics used with the UDI are included, the implant card information cannot be generically the same for each device but that it will have sections that are specific to individual devices. Sure this may initially create some logistical headaches for keeping track that the implant cards don’t get mixed up in situations where the devices are being manufactured, but this creates a level of accountability that is designed for the ultimate safety of the end patient.
Section 1. Sub-section B
Sub-section B contains the warning information of the device. The first part is pretty self-explanatory as meaning literally what is stated “any warnings” and “precautions”. It is the next part that I do not interpret literally. Where it says “measures to be taken by the patient or a healthcare professional with regard to reciprocal interference with reasonably foreseeable external influences, medical examinations or environmental conditions”.
If I were the manufacture of an implantable medical device, I would most definitely include measures to be taken by the patient as well as measures to be taken by a healthcare professional. There are a couple of spots that use the word ‘or’, and if it were me, I would read it ‘as well as’.
I say that for a few reasons. One is that without explicit clarification of a governing body as exactly what a silly little word like that is intended to me, this creates an area that is open for debate. Does that ‘or’ mean that at least one of those needs to be included and the rest can be excluded?
As one who likes to err on the side of caution, if you have the information available, why would you not provide it? By going above and beyond not only demonstrates your goodwill but also avoids hang-ups where an auditor might not agree with how you viewed the requirement, and you end up with a nonconformity, or in the same situation with an incident investigator. Ink is cheap; liabilities are expensive.
Section 1. Sub-section C, and Sub-section D.
These two subsections are relatively short and straight forward.
“(c) any information about the expected lifetime of the device and any necessary follow-up;
How long can the user expect your device to last once it has been implanted? I there any maintenance they should be performed? Perhaps once a year, a physician needs to double-check the device placement?
(d) any other information to ensure the safe use of the device by the patient, including the information in point (u) of Section 23.4 of Annex I.”
The rest of Section 1. Of Article 18.
“The information referred to in the first subparagraph shall be provided, to make it available to the particular patient who has been implanted with the device, by any means that allow rapid access to that information and shall be stated in the language(s) determined by the concerned Member State. The information shall be written in a way that is readily understood by a layperson and shall be updated where appropriate. Updates of the information shall be made available to the patient via the website mentioned in point (a) of the first subparagraph.
Also, the manufacturer shall provide the information referred to in point (a) of the first subparagraph on an implant card delivered with the device.”
At the end of this section, it provides a little bit more information about the purpose of the article but also lays out some guidelines for how to make the required information available.
I specifically mentioned earlier that having the information slapped on a website is not enough by itself. The text states, “any means that allow rapid access to that information”. Certainly, available on the internet is a means that allows rapid access, and it is if you have internet. Using a web-based approach like that is assuming that all the possible patients all have the technology and budget to reach the information. This means that every single possible patient needs a means to access the internet, and the money to pay for internet access. Also, being able to simply access the information rapidly isn’t necessarily providing the information “together with the device” as required.
You also need to have a conversation with your notified body and determine what languages are required by the member state in which your device is sold. It does not do the patient much good if they do not understand the language in which the information is being presented. It also needs to be presented in easy to understand terms, not in technical jargon.
Updates, unlike the initial presentation of information, needs to be included on your website. Specifically, the website that was included in the implant card given to the patient.
Section 2. of Article 18
Unlike what we saw in Section 1. Section 2. Outlines requirements for the health institutions and not the manufacturer. More specifically, Section 2. Requires member states to require health institutions to perform actions.
This section makes health institutions provide the same information that manufacturers had to provide to patients who have been implanted with a device, with the same stipulations as to how the information is provided. However, it also includes the health institution to include their identity on the implant card as well.
Member States shall require health institutions to make the information referred to in paragraph 1 available, by any means that allow rapid access to that information, to any patients who have been implanted with the device, together with the implant card, which shall bear their identity.
Section 3 of Article 18 is the list of exempted implants, exempted devices are:
Sutures
Staples
Dental Fillings
Dental Braces
Tooth Crowns
Screws
Wedges
Plates
Wires
Pins
Clips
This is not an exhaustive list and can change with time at the discretion of the Commission. What it has done is taken implanted devices and exempted some of the most common and widely used ones. Thankfully so too, imagine if every staple needed an implant card to be presented to the receiving patient with individual batch and identifying numbers. Then coordinate the effort with a health institution so that the card also bears their identification as well. This would quickly become exhaustive.
The following implants shall be exempted from the obligations laid down in this Article: sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors. The Commission is empowered to adopt delegated acts in accordance with Article 115 to amend this list by adding other types of implants to it or by removing implants therefrom.”


 Matthew Walker – QMS, Risk Management, Usability Testing, Cybersecurity
Matthew Walker – QMS, Risk Management, Usability Testing, Cybersecurity