This 510k software documentation webinar defines the new FDA eSTAR documentation required for software and cybersecurity.
510(k) Software Documentation Webinar – Free
This webinar was created in 2019, but Rob Packard is presenting an updated version on March 7, 2024, to address the new requirements of the FDA eSTAR template (v5.1) in the software and cybersecurity sections. If you cannot attend the live session, we will record it and provide you with a download link if you fill out the form below.
When is the 510k Software Documentation Webinar?
The live webinar is scheduled for March 7, 2024 @ 10:30 am ET. If you fill out the form above before the live presentation, you will receive a link to participate in the live webinar via Zoom. For SaMD and SiMD, modify your design plan to integrate 510(k) software documentation requirements and cybersecurity documentation requirements. Specifically, the new FDA eSTAR requires 10 different document types to be attached for software documentation and 12 different document types to be attached for cybersecurity. The most important thing to consider is which documents must be created before you start coding and which are created only after you are done coding. The documents can and will be revised and edited during the software development. Still, your project will not be successful if you don’t build the proper foundation for the project by creating a few critical documents before your team starts coding. This webinar is also applicable to software as a medical device (SaMD).
510(k) Software Documentation Webinar
Risk Documentation Level – The FDA no longer uses the term “Level of Concern” and the level of software risk documentation were simplified to “Basic Documentation” and “Enhanced Documentation.” You only need to justify why your software does not fall within the criteria for “Enhanced Documentation.” We also provide a template for this in SYS-044. The documentation requirements for software and cybersecurity are explained further in our YouTube video for v5.0 software documentation.
-
- Software Description – A summary overview of the features and software operating environment.
- Device Hazard Analysis – Tabular description of identified hardware and software hazards, including severity assessment and mitigations.
- Software Requirement Specification (SRS) – This documents the software requirements about hardware requirements, programming language requirements, interface requirements, software performance, and functional requirements.
- Architecture Design Chart – Detailed depiction of functional units and software modules. It may include state diagrams as well as flow charts.
- Software Design Specification (SDS) – The SDS describes how the requirements in the SRS are implemented. The information presented in the SDS should be sufficient to ensure that the work performed by the software engineers who created the Software Device was clear and unambiguous, with minimal ad hoc design decisions. The SDS may reference other documents, such as detailed software specifications.
- Traceability Analysis – Traceability among requirements, specifications, identified hazards and mitigations, and Verification and Validation testing.
- Software Development Environment Description – Summary of software life cycle development plan, including a summary of the configuration management and maintenance configuration activities.
- Verification and Validation Documentation – Description of software verification and validation activities at the unit, integration, and system level. System-level test protocol, including pass/fail criteria and test results.
- Revision Level History – Revision history log, including release version number and date.
- Unresolved Anomalies (Bugs or Defects) – List of remaining software anomalies annotated with an explanation of the impact on safety or effectiveness, including operator usage and human factors.
- Cybersecurity – The process of preventing unauthorized access, modification, misuse or denial of use, or the unauthorized use of information that is stored, accessed, or transferred from a medical device to an external recipient.
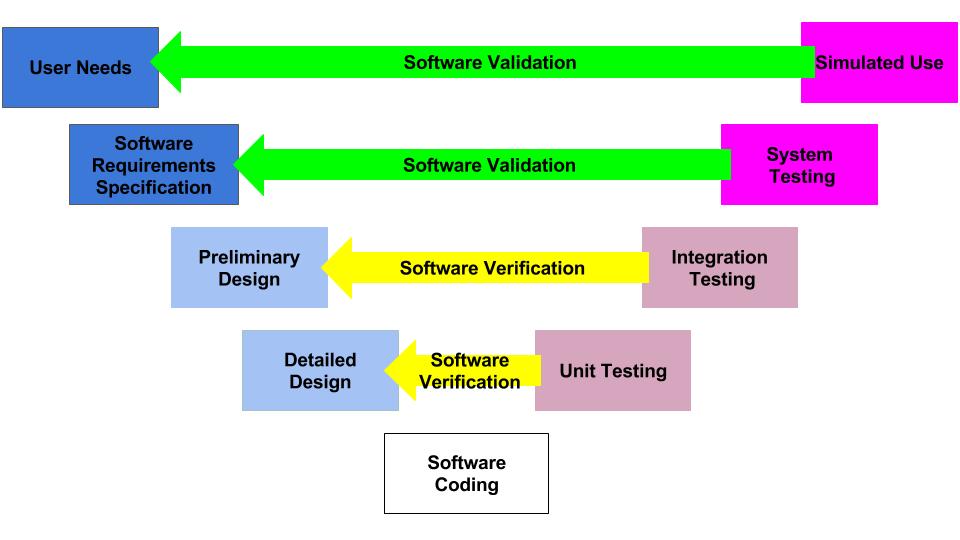
Our procedure for software validation (SYS-044) is available:
Or click on the image below for other procedures:
")
Additional Resources for 510k submissions
If you would like additional training on 510k submissions or you would like to access Medical Device Academy’s templates, you can purchase all of our templates and 510k webinars on our 510k course webpage.
About Your Instructor
 Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.
Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.