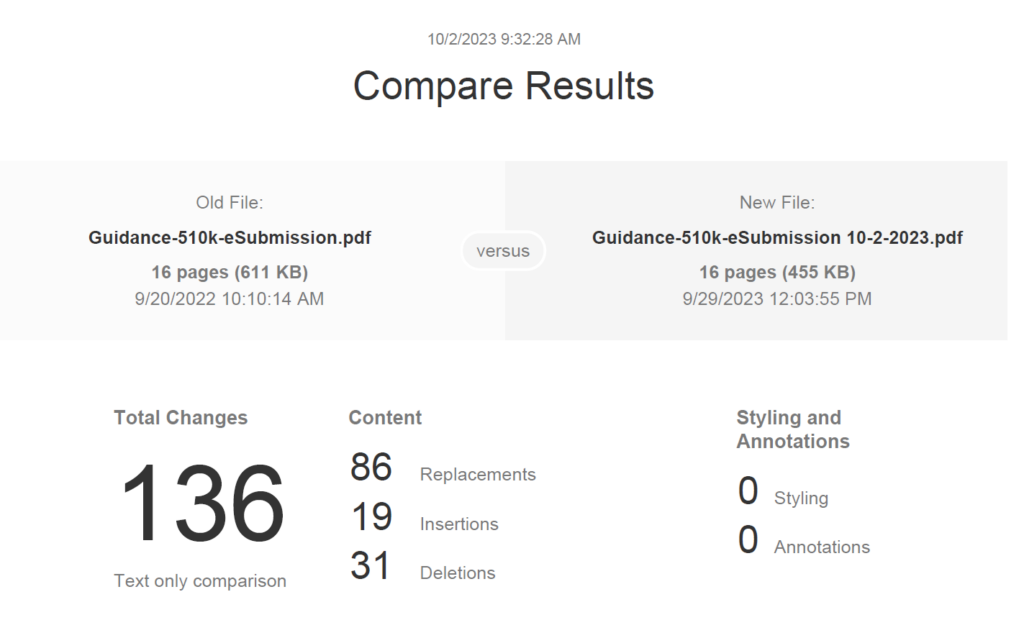
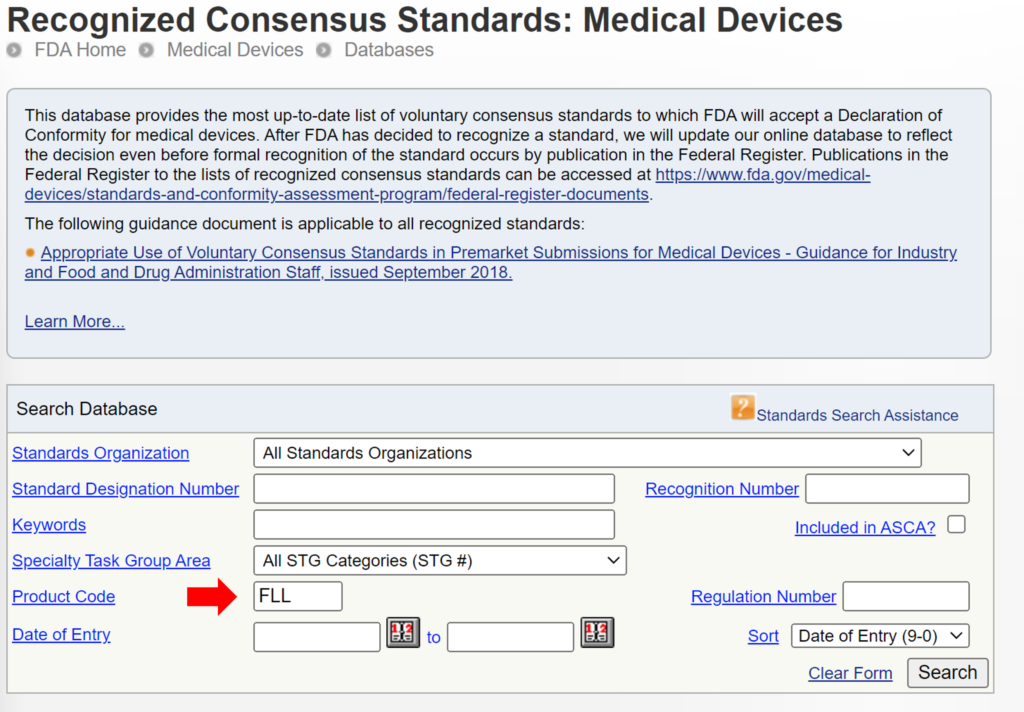
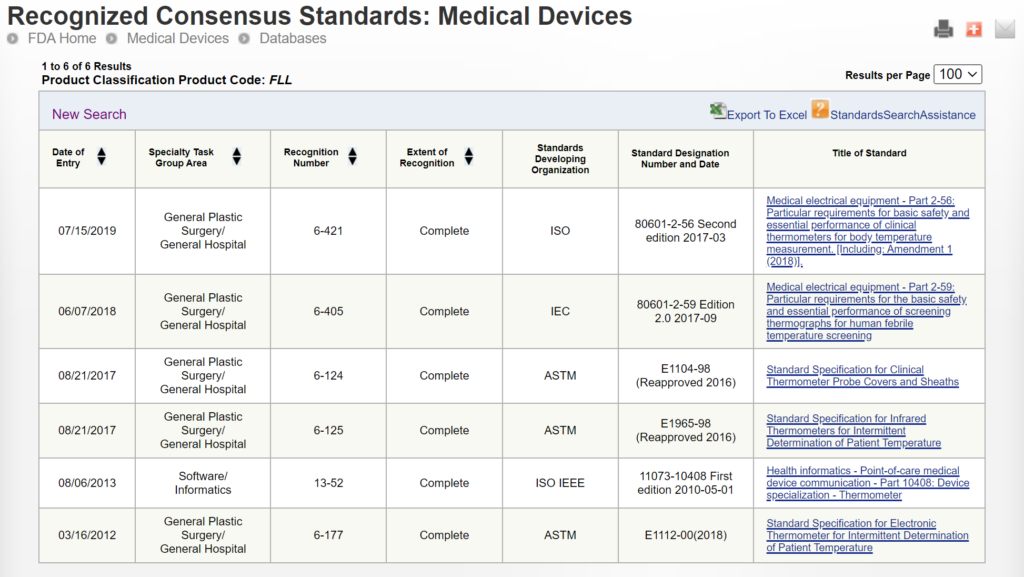
In the eSTAR and PreSTAR, the FDA inquires whether a request for designation (RFD) is associated with your device.
What is a request for designation (RFD)?
A request for designation is a formal request to the Office of Combination Products (OCP), where you request that OCP assign the agency division that will have jurisdiction over a combination product. In 21 CFR 3.7, the FDA outlines the information required in an RFD submission. The FDA encourages RFD submitters to review the agency’s guidance prior to submitting an RFD. It irritates the FDA when you don’t read the guidance and ask questions that are clearly answered within it. Read the guidance first (we provided links below).
What is a combination product?
Before you submit a request for designation, you need to understand what a combination product is. The term combination product includes:
A product comprised of two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity;
Two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products;
A drug, device, or biological product packaged separately that, according to its investigational plan or proposed labeling, is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of the approved product would need to be changed, e.g., to reflect a change in the intended use, dosage form, strength, route of administration, 9or significant change in dose; or
Any investigational drug, device, or biological product packaged separately that, according to its proposed labeling, is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
Information regarding the drug/biologic constituent part of the combination product may be needed and accounted for throughout the various sections of your premarket submission. In addition, as described in Product Stability documentation, medicinal substance refers to the drug/biologic constituent part of the combination product as defined in 21 CFR 3.2(e).
How do you write a request for designation (RFD)?
We recommend that you always start with a pre-request for designation (pre-RFD). Once you have feedback from the FDA, then you will be ready to write your request for designation (RFD). The FDA published two guidance documents related to RFDs:
the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and/or
whether CBER, CDER, or CDRH will regulate the product if it is a non-combination product, or
which of those Agency Centers will have primary jurisdiction for a premarket submission of a combination product.
The FDA’s target review time is 60 days for providing the information requested, but a pre-RFD is not a tracked metric with budget impact. Therefore, you should set expectations with your senior management team and investors at approximately 90 days–just like the 513(g) submissions.
21 CFR §3.7 – Request for Designation (copied from eCFR)
(a) Who should file: the sponsor of:
Any combination product the sponsor believes is not covered by an intercenter agreement; or
Any product where the agency component with primary jurisdiction is unclear or in dispute.
(b) When to file: a sponsor should file a request for designation before filing any application for premarket review, whether an application for marketing approval or a required investigational notice. Sponsors are encouraged to file a request for designation as soon as there is sufficient information for the agency to make a determination.
(c) What to file: an original and two copies of the request for designation must be filed. The request for designation must not exceed 15 pages, including attachments, and must set forth:
The identity of the sponsor, including company name and address, establishment registration number, company contact person and telephone number.
A description of the product, including:
Classification, name of the product and all component products, if applicable;
Common, generic, or usual name of the product and all component products;
Proprietary name of the product;
Identification of any component of the product that already has received premarket approval, is marketed as not being subject to premarket approval, or has received an investigational exemption, the identity of the sponsors, and the status of any discussions or agreements between the sponsors regarding the use of this product as a component of a new combination product.
Chemical, physical, or biological composition;
Status and brief reports of the results of developmental work, including animal testing;
Description of the manufacturing processes, including the sources of all components;
Proposed use or indications;
Description of all known modes of action, the sponsor’s identification of the single mode of action that provides the most important therapeutic action of the product, and the basis for that determination.
Schedule and duration of use;
Dose and route of administration of drug or biologic;
Description of related products, including the regulatory status of those related products; and
Any other relevant information.
The sponsor’s recommendation as to which agency component should have primary jurisdiction based on the mode of action that provides the most important therapeutic action of the combination product. If the sponsor cannot determine with reasonable certainty which mode of action provides the most important therapeutic action of the combination product, the sponsor’s recommendation must be based on the assignment algorithm set forth in § 3.4(b) and an assessment of the assignment of other combination products the sponsor wishes FDA to consider during the assignment of its combination product.
(d) Where to file: all communications pursuant to this subpart shall be addressed to the attention of the product jurisdiction officer. Such a request, in its mailing cover should be plainly marked “Request for Designation.” Concurrent submissions of electronic copies of Requests for Designation may be addressed to combination@fda.gov.
This blog provides a deep dive into the newest version of the FDA eSTAR, version 5.5, released on February 12, 2025.
Why did the FDA release the new eSTAR version as v5.5 instead of v6.0?
A major version update consists of policy changes, regulatory changes, or major changes to the template and will be denoted by a major version number increment (e.g. 5.4 to 6.0). A minor version update will consist of other changes and will be denoted by a minor version number increment (e.g. 5.4 to 5.5). If there are policy or regulatory changes, a new major version of the eSTAR is made before the implementation date, and the previous version of the eSTAR is removed. As an example, the FDA updated v4.3 to v4.4 to enable PMA content, updates to the international pilot of the eSTAR with Health Canada, and implementation of cybersecurity documentation requirements are considered major changes that trigger the need for a major version update (i.e., 5.0) instead of a minor version update (i.e., 4.4). These changes apply to the IVD eSTAR and the non-IVD eSTAR. If you are generally unfamiliar with the FDA eSTAR, please visit our 510k course page.
What is the deadline for using v5.5?
Version 5.4 of the FDA eSTAR, both the nIVD and IVD versions, may continue to be used until v6.0 is eventually released. In fact, any v5.x may be used until v6.0 is released. Any submissions that are submitted with an expired version (v4.x) of the eSTAR will be rejected. If you have already uploaded information to an older version of the template, you will need to scroll to the bottom of the eSTAR and export the data to an HTML file. Then you import the HTML file into the newer version of the eSTAR. Any attachments you made to the older version of the template will not be exported, and you will have to attach all of the attachments to the new template.
PMA content is enabled in the new FDA eSTAR
Previous versions of the FDA eSTAR included the functionality for premarket approval (PMA) submissions, but in version 5.0 the FDA finally enabled this functionality. 510k submissions have three types: 1) Traditional, 2) Abbreviated, and 3) Special. PMA submissions also have different types. There are two types of PMA submissions for a new device: traditional and modular. Unfortunately, the FDA eSTAR is not intended for PMAs using the modular approach. For Class 3 devices, the FDA has more stringent controls over changes than Class 1 and 2 devices. Therefore, a PMA supplement is required for the following types of changes to PMA-approved devices:
new indications for use;
labeling changes;
facility changes for manufacturing or packaging;
changes in manufacturing methods;
changes in quality control procedures;
changes in sterilization procedures;
changes in packaging;
changes in the performance or design specifications, and
extension of the expiration date.
There are several types of PMA supplements, but only three types of supplements can use the FDA eSTAR: 1) Panel-Track, 2) 180-Day, and 3) Real Time. To determine which type of PMA supplement you should use, the FDA published guidance for modifications to devices subject to the premarket approval process.
PMA Content
The following sections in the FDA eSTAR are specific to PMA submission content requirements:
Quality Management System Information
Facility Information
Post-Market Study (PMS) Plans
Attach an exclusion statement, or an Environmental Assessment Report in accordance with 21 CFR 814.20(b)(11)
Health Canada is conducting a pilot with the FDA eSTAR
Health Canada’s FDA eSTAR pilot is now full with a total of 10 participants (originally only 9 were planned). The pilot will test the use of eSTAR for applications submitted to Health Canada. The results of the pilot should be complete soon, and then we expect an extension of the pilot to a broader number of applicants. We heard rumors that the HC eSTAR was overly complicated. Hopefully, future versions are simplified.
Were there any changes to the EMC testing section?
EMC Labeling questions were consolidated into a single question instead of four because only one citation is usually provided in this section. A copy of the older version is provided below.
The updated version 5.0 is shown below and has only one question, but the help text was changed.
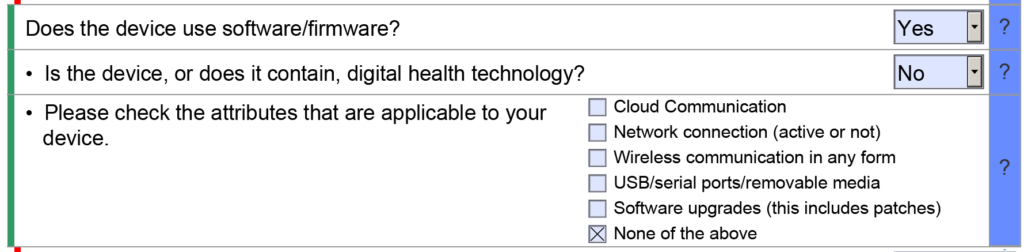
Does the FDA eSTAR now require more cybersecurity documentation?
We have updated our cybersecurity work instruction (WI-007) to address the updated FDA guidance for cybersecurity documentation. The revisions were completed earlier this month, and you can purchase the updated templates on our website. We have also been telling our subscribers to anticipate a significant revision to the FDA eSTAR template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device:
risk management – report (attach)
risk management – threat model (attach)
list of threat methodology (text box)
verification that the threat model documentation includes (yes/no dropdown):
global system view
Multi-patient harm view
Updateability/patchability view
Security use case views
cybersecurity risk assessment (attach)
page numbers where methodology and acceptance criteria are documented (text box)
verification that the risk assessment avoids using probability for the likelihood assessment and use exploitability instead (yes/no dropdown)
software bill of materials or SBOM (attach)
software level of support and end-of-support date for each software component (attach)
operating system and version used (text box)
safety and security assessment of vulnerabilities (attach)
assessment of any unresolved anomalies (attach)
data from monitoring cybersecurity metrics (attach)
information about security controls (attach)
page numbers where each security control is addressed (text box):
Authentication controls
Authorization controls
Cryptography controls
Code, data, and execution integrity controls
Confidentiality controls
Event detection and logging controls
Resiliency and recovery controls
Firmware and software update controls
architecture views (attach)
cybersecurity testing (attach)
page numbers where cybersecurity labeling is provided (text box)
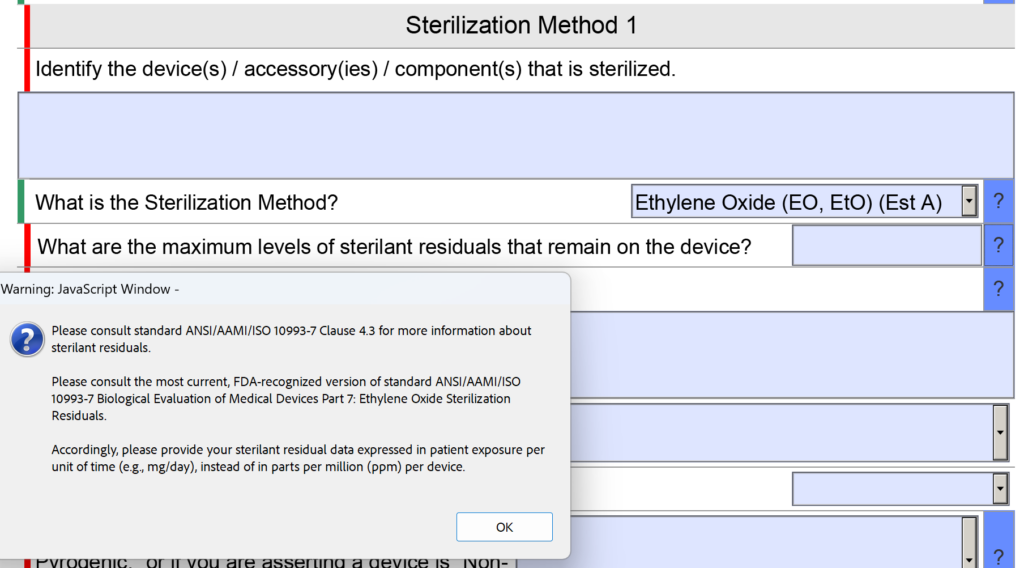
Sterility section changes include an updated question on EO residuals
In the sterility section of the FDA eSTAR there was a question about sterilant residues. Specifically, the question was “What are the maximum levels of sterilant residual that remain on the device?” The space provided for entering the information was small as well.
Now the question is reworded to: “What are the maximum levels of sterilant residuals that remain on the device, and what is your explanation for why those levels are acceptable for the device type and the expected duration of patient contact?” No change was made to the help text for this question.
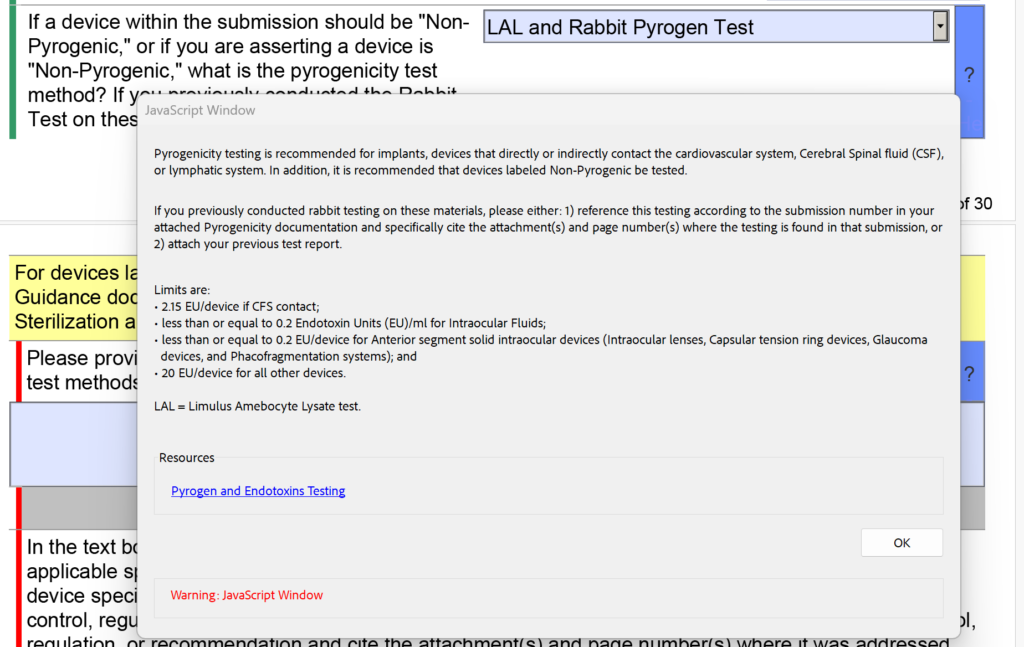
In addition to the changes in the sterility section regarding EO residuals, the FDA also modified the dropdown menu and the help text for pyrogenicity testing. There were options for “LAL” and “Rabbit Test” separately, but now these are combined into “LAL and Rabbit Pyrogen Test.” In addition, the following help text was added: “If you previously conducted rabbit testing on these materials, please either: 1) reference this testing according to the submission number in your attached Pyrogenicity documentation and specifically cite the attachment(s) and page number(s) where the testing is found in that submission, or 2) attach your previous test report.”
What is the deadline for using v5.0?
Many clients say that they get an error message when they try to open the FDA eSTAR template. This is because they are opening the eSTAR from a PDF viewer instead of Adobe Acrobat Pro.
Some people want to save money by using the free Adobe Acrobat Reader software instead, but this will not allow you to complete the eSTAR properly. Therefore, the FDA added a Popup message if Adobe Acrobat Reader is used.
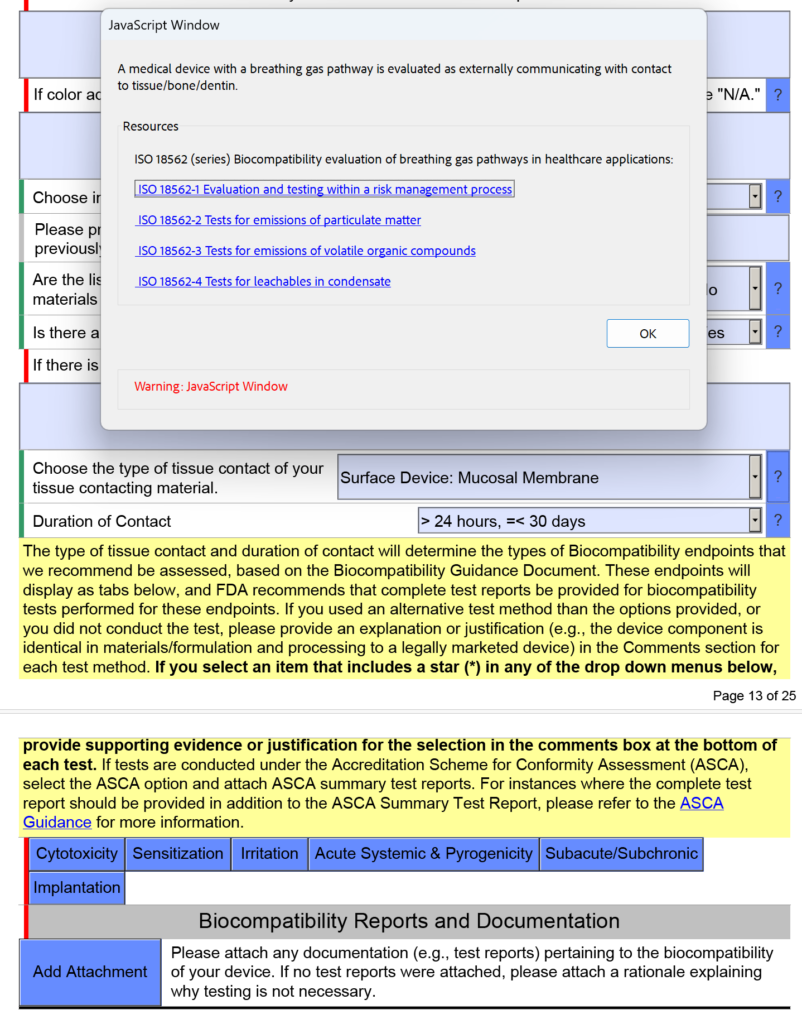
How are devices with a breathing gas pathway evaluated for biocompatibility?
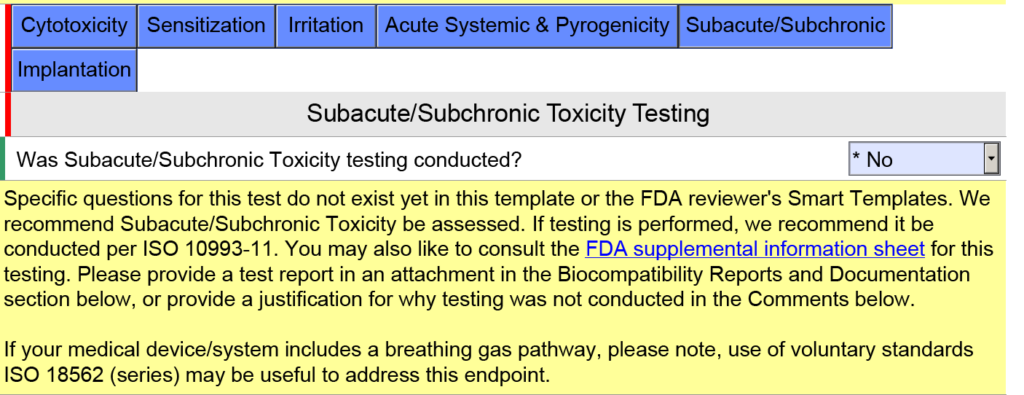
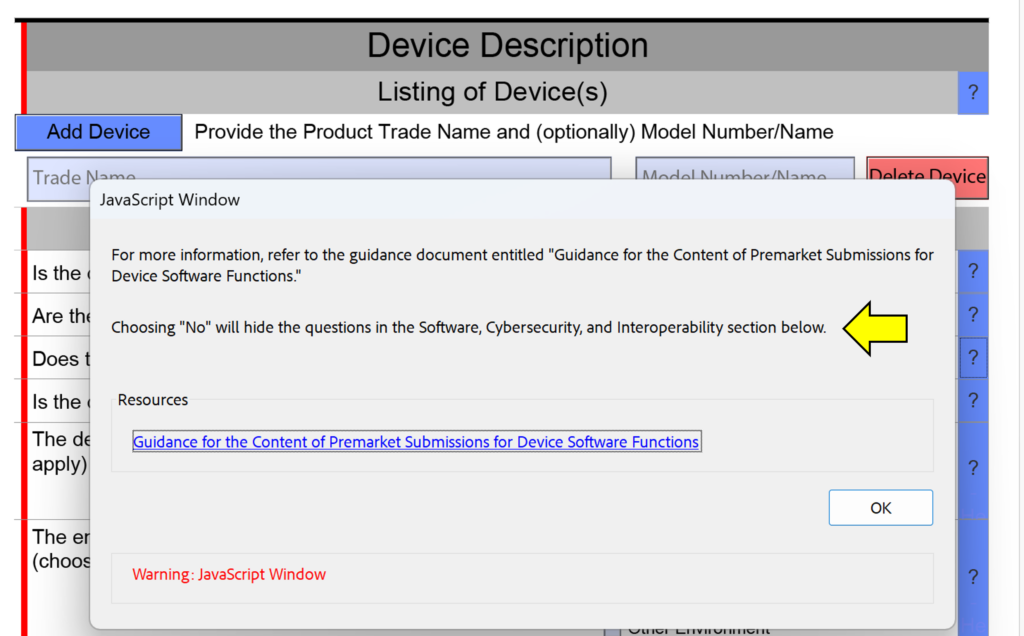
In the screen capture below, I have intentionally selected “Surface Device: Mucosal Membrane” as the type of tissue contact for a breathing gas pathway device because the device will have a mouthpiece placed in your mouth (i.e., mucosal membrane). This is a common mistake. In version 5.0 of the FDA eSTAR, the FDA clarifies that these devices should be evaluated as “externally communicating” and the tissue contact is “tissue/bone/dentin.” Specifically, the tissue contact is the lungs. For this reason, the FDA added the help text shown below in the JavaScript Window regarding the applicability of ISO 18562-1, -2, -3, and -4.
Additional questions and guidance will appear when you click on the individual blue boxes shown above. For the blue box labeled “Subacute/Subchronic,” you will find additional help text regarding the ISO 18562 standards. Similar help text is found when you click the blue box labeled “Acute Systemic & Pyrogenicity.”
What is a cross-section change reminder?
One of the minor changes made in this FDA eSTAR version is the addition of “cross-section change reminders” to the help text in the device description section. This is not meant to help you avoid answering questions in your submission, because if you are missing a section of the submission because you answered “No” instead of “Yes” the FDA reviewer will identify this error during the Technical Review process. This will result in your submission being placed on hold and the review time clock will be reset to zero days when you resubmit with the corrections made. The screen capture below shows an example of one of these cross-section change reminders.
What changes were made to the clinical testing section of the FDA eSTAR?
The clinical testing section will now display when using PDF-XChange Editor, but we recommend only using Adobe Acrobat Pro to edit the FDA eSTAR. This change is a bug fix, and it is specific to the nIVD eSTAR. The IVD eSTAR and the nIVD eSTAR both include a clinical testing section within the performance testing section, but the performance testing section is found in the FDA eSTAR before the electrical safety and EMC testing section, while the performance testing section is found after the electrical safety and EMC testing section. If your company is planning to submit clinical data in a future FDA submission, we have the following recommendations:
conduct a pre-submission teleconference to ask questions about your clinical study protocol before IRB submission or ethics review board submission
before you submit the pre-sub meeting request, look at what general clinical information the FDA wants for a De Novo or PMA submission in the FDA eSTAR
Note: The clinical section shown above is only found in the FDA eSTAR if you select a De Novo or PMA submission. If you submit a 510k submission with clinical data, the clinical section will be abbreviated as shown below.
If you are a third-party or hospital reprocessor, learn how to prepare an FDA eSTAR 510(k) submission for reprocessed single-use devices.
Why is there so much interest in reprocessed single-use medical devices?
With increasing pressures on the medical device industry to make healthcare more affordable, there has been a push to reprocess and reuse single-use devices. Reprocessors obtain used devices from healthcare facilities. The reprocessors clean, process, resterilize, repackage, and relabel devices. Reprocessors must obtain FDA 510(k) clearance by demonstrating that the safety and effectiveness of the reprocessed device are substantially equivalent to the single-use device produced by the original equipment manufacturer (OEM). The FDA created a FAQ document for single-use devices, and three guidance documents were published:
Why do reprocessors have difficulty preparing an FDA eSTAR for reprocessed single-use devices?
Obtaining 510(k) clearance for a device your company did not design can be challenging because the reprocessor doesn’t have access to all of the required design and manufacturing information. The following sections of the FDA eSTAR submission pose unique challenges for reprocessed single-use devices:
Labeling – What should and should not be included in the reprocessed device labeling
Biocompatibility – How to identify the materials, and determine what biocompatibility testing needs to be done
Performance Testing – Strategies for determining appropriate performance testing
Labeling Section of the FDA eSTAR for reprocessed devices
Labeling of reprocessed devices consists of the instructions for use and the packaging label(s). Device package labeling may also direct the user to both the reprocessor’s IFU and the OEM’s IFU. If you are referencing the OEM’s IFU, it is also important to include the OEM’s model number. Instructions for use should consist of:
Indications for use, which must be equivalent to the OEM indications.
All of the necessary warnings and cautions and basic operating instructions needed to operate the device safely.
The instructions for use may also instruct the user to reference the OEM instructions for use for additional information.
Instructions on the handling of the device after use, with the likelihood that the device will be returned to the reprocessor to repeat the cycle.
Biocompatibility Section of the FDA eSTAR
Biocompatibility data is more challenging to provide if you replace or modify original components. If reprocessing does not modify the OEM device whatsoever, you can claim that the materials are identical to the OEM device. Therefore, the reprocessed device does not require biocompatibility testing. However, the reprocessor still needs to evaluate the biological risks associated with the reprocessing of the device by testing for cleaning and sterilization residuals. This involves testing for cleaning agent residuals and EO residual testing (ISO 10993-7), if applicable. If applicable, this involves testing for cleaning agent residuals and EO residual testing (ISO 10993-7)
If you replace any of the components during reprocessing with a new component that is identical in dimension and material to the OEM component, minimal biocompatibility testing will be required. If the exact material used by the OEM is unknown, reprocessors can perform material identification testing to determine the material used, and then create the replacement part out of the same material.
If you modify or replace any patient-contacting components on the device such as lubricants, insulation, etc., with components that are different from the OEM, then you will need to perform additional biocompatibility testing to prove that the new or modified material is biocompatible. This testing will depend on the duration of contact and where will the material contact the patient. The new material will also need to be listed in your device description and Section 15 of your 510(k) submission.
Performance Testing Section of the FDA eSTAR
There are three primary sources for identifying performance testing requirements of reprocessed devices:
OEM Testing listed in the OEM 510(k) submission
Predicate Testing listed by another reprocessor of an equivalent device
Product Standards listed under the product classification code for the reprocessed device or the OEM device
You should reference a predicate device that has been reprocessed and the OEM device to identify performance testing. Some testing is specific to the functional performance of the device. For these tests, you need to compare performance side-by-side against the OEM. Another testing is specific to reprocessing; you will reference the predicate device. Sources of information regarding the required tests for each of these devices can be found in the 510(k) summaries of the respective devices. If possible, it’s helpful to select a predicate that has a redacted 510(k) available on the FDA’s website. If a redacted 510(K) is not readily available, you may request a redacted copy through the Freedom of Information Act online. A redacted copy of the OEM 510(k) is also helpful. It’s helpful to select a predicate with a redacted 510(k) available on the FDA’s website if possible
If testing information is not as readily available in the 510(k) summary, you will determine the essential performance functions of the device, and design tests to evaluate and compare the OEM device and the reprocessed device for those functionalities. Some devices have specific standards for their design and/or testing. To determine if the reprocessed device has any applicable standards, you should search the product code of the reprocessed device and the product code of the OEM device, if they are different, in the FDA product classification database. The search results will list recognized standards applicable to the reprocessed device.
Additional tests that may be needed to validate reprocessing include residual protein, residual carbohydrates, and the presence of hemoglobin. These tests ensure that all biological material from previous use is removed. If you are not performing biocompatibility testing on the reprocessed device, you must do a chemical test to ensure no residual detergent or cleaning residues remain on the device. You must also determine how many reprocessing cycles the device can survive before performance degradation. This can be done by repeating simulated use, reprocessing, and performance testing until a statistically relevant decrease in the performance of the device is observed.
If you have additional questions regarding preparing your 510(k) submission, please visit our Contact Us webpage to schedule a meeting with Lindsey Walker or Rob Packard.
The author discusses his personal experience with quality and regulatory training and shares his secrets for better instructor engagement.
What is instructor engagement?
Instructor engagement is a term that describes actions taken by the instructor to involve their class, whether the training is on-line or in person. Instructor engagement includes six basic elements:
Training content and format
Verbal communication
Non-verbal communication
Training environment
Audience
Audience involvement
Any one of the above six elements can ruin a training class, but a great instructor can compensate for weaknesses in any one area by taking advantage of the other elements.
To be successful, you need to hook your audience in the first ten seconds
It is crucial to engage your audience in the first ten seconds. When the audience is live, if you don’t engage them immediately they will find something on their phone to distract them. If the audience is on-line, they will swipe to the next video in their feed. Toastmasters suggests beginning your presentation by using one of five methods for hooking your audience:
Tell a story
Make a bold statement
Ask a question
Get the audience to laugh
Ask the audience to visualize something
Consequences of poor engagement
If an instructor does not engage students, the best case scenario is that the training will not be effective. In other words, the class will not learn the material being taught or they will retain the information for less than 24 hours. For the student, their time and money was wasted. For the instructor, they will feel exhausted at the end of the training and they will have trouble finding future training jobs.
Results of good instructor engagement
If a class is engaged in training they will learn the material, retain the information in their long-term memory, and the will recommend the instructor to other people that are interested in learning about the topic. For the student, their time and money was well spent. For the instructor, they will feel energized at the end of the training and students will come up to them at the end of the training asking for a business card and to discuss future training opportunities.
Where can you find an examples of good and bad instructor engagement?
When I first wrote a blog on this topic it was 2012 and there were very few blogs and almost no videos dedicated to quality systems or regulatory affairs. Twelve years later, almost nobody reads blogs and there are multiple competitors that publish new videos weekly. The primary channel for watching videos is YouTube, but YouTubers are simultaneously live-streaming on YouTube, LinkedIn, and Facebook. These channels are the best place to find examples of good and bad instructor engagement. You will probably have a strong opinion about the quality of the speaker in the first ten seconds, but the algorithms that guide your surfing of these platforms will automatically steer your viewing to the best videos. These videos are not considered to be the best solely on content and format. The amount of audience engagement is the biggest driver. The algorithm recommends videos based upon the percentage of audience retention, the number of comments by viewers, and the number of people that share the link to your video.
Why doesn’t Dr. Shulman’s video rank higher in the algorithm?
The video I embedded in this post has only 554 views currently, but it has been posted on YouTube almost six years. Why doesn’t this video rank higher and get recommended by YouTube to more people? Because there is no involvement of the original audience or the current YouTube audience. Despite the talented speaker and the use of video with clear PPT slides, there are no comments on YouTube. The content and format is good, and the verbal communication is good. However, RAPS has Dr. Shulman standing behind a podium at the event so the non-verbal communication is not as strong as it could be. In addition, large conferences are typically one of the weakest environments for encouraging instructor engagement with the audience. The video could receive much higher rankings if the original audience was involved in the video as well. If the question and answer session were included, that might have helped. RAPS could also improve the video’s performance by adding more details to the description of the video. Finally, the performance of the video is impacted by the number of subscribers to the RAPS YouTube channel and the engagement of those subscribers. To demonstrate this, I will add a comment to the video. That should cause a small increase in engagement and viewers.
How can you improve your internal quality system training?
Anyone can read and understand a procedure, but this is the least effective method of training people. You could also have employees watch a training video, but quality assurance and regulatory affairs are among the most boring topics on planet earth. Most of the training out there is “Blah, blah, blah…” and “Death by PowerPoint.” Instructor engagement for that type of training is poor, and it could get you fired. Don’t read your slides, don’t turn your back on the audience (or they’ll attack), and PLEASE don’t ever ask someone to read the definition of nonconformity out loud to the rest of the group. Inspire and engage the class. You need to get your audience to pay attention, ask questions, and share their own thoughts out loud. For example, instead of using a PowerPoint, try displaying the actual procedure and ask an audience member to find each of the points you are teaching in the procedure. You could even teach them a cool search tool (i.e., CTRL + F) to find the content. You might even adding a symbol to the procedure to help them find those requirements.
Nine ways to improve your own instructor engagement
If you are hiring a consultant to help you with quality and regulatory training, then you certainly want to hire an expert. However, it is more important that the speaker is engaging. Knowing this fact, you could try improving your own presentation skills to achieve higher instructor engagement for a lot less money. I’m six-foot, six inches tall, and I have a loud booming voice. My mother has red hair, and she was an opera singer. I’ve got the voice to fill any auditorium and stage presence to match. But you don’t have to be big, tall, or loud to capture the attention of your audience. Here are my top nine ways to improve instructor engagement:
Practice vocal variety
Move, don’t stay stationary
Ask the audience questions
Use anecdotes, case studies, and stories
Try using props
Take breaks
Plan a surprise
Force feed the audience legal stimulants
Give students homework
What is vocal variety, how does it impact instructor engagement?
Vocal variety is more than just the volume of the speaker. Vocal variety consists of pitch, tone, volume, and pace. Generally I speak too fast and my voice is very loud. Therefore, if I want to emphasize a point I can exercise two changes in my voice to immediately capture the attention of students: 1) speak softly, and 2) speak slowly. Another approach I have used, is to speak slowly and repeat myself, but the most dynamic way to get your audience’s attention is to stop speaking for a few seconds. Silence is powerful.
Many people struggle to understand how to vary their pitch, but they’re overthinking it. When we ask a question, we raise the pitch of our voice slightly at the end. This upward inflexion of our voice signals to listeners that we are asking a question. For example, if you repeat the last one to three words in the other person’s sentence, and you say this with an upward inflection, the listener will perceive that you are asking a question to better understand what they mean. This is much more effective than asking “what do you mean?” For example, if a student in the class says “Is it enough if we perform annual reviews?” As the instructor you can employ the technique of mirroring by asking, “Annual reviews?” This will encourage the other person to elaborate on what they meant by “annual reviews.” This technique ensures that you know what the student meant, and it gives you time to consider what they said before you respond. This is also a strategy recommended by expert negotiators.
Movement is attractive
Most of our brain power is dedicated to processing what we see–not what we hear. Therefore, listeners are more likely to notice when you move. You can jolt your audience awake simply by stepping out from behind a podium or changing your movement pattern (e.g., occasionally moving forward instead of pacing side to side). Movement also includes body language. You can modify your posture, stance, and position in front of the audience to communicate information non-verbally. You can use gestures to communicate non-verbally or you can use facial expressions. For example, if you frown and shrug your shoulders, what does that mean?
Questions are essential for instructor engagement
No matter how smooth and eloquent your voice is, nobody wants to hear only you speak. This is one of the reasons conferences have multiple speakers. However, during a single presentation you can get students to participate and present some of the information by asking them questions. There are a few techniques that help get the audience to speak more. Instead of asking a question to the group as a whole, try pointing to a specific person, ask them their name, and then ask them a question. Second, in order to “break the ice” at the end of your presentation, have some “seed questions” prepared. If you are conducting a webinar, seed questions can be read by you. In a live presentation, you can give a list of seed questions to your host or friends in the audience. Finally, you can also begin your training with a question (i.e., Toastmaster hook #3 above).
Tell a story and make a point
For each teaching point you should consider using an anecdote, case study, or a related story. The audience will want to know how the story ends, and they are more likely to remember the story. If you haven’t conducted more than 1,000 audits, traveled all over the world, or have more than 25 years of experience–don’t despair. You can always interview other people on any topic to get their stories. If you are looking for tips on how to construct a story, Toastmasters comes to my rescue again. Here’s their recommended six-part structure for the Hero’s Journey:
Setup
Inciting incident
Progressive complications
Insight
Climax and resolution
Lesson
What kind of prop can you use for quality and regulatory?
In one of the paragraphs below, I mention a simple prop that you can use for training–product samples. I did this in one of our live-streaming YouTube videos where I explained how to review medical device labeling. But you can use other things in your environment. For example, the first public speaking course I ever had was taught by a man with Polio that used crutches. He used one of his crutches as a prop to demonstrate how he looked over a fence.
How often should you take breaks?
Students cannot maintain a state of alertness and attention indefinitely. Your body naturally cycles between higher and lower alertness every 90 minutes. After 90 minutes it becomes harder to focus and you need to take a break. If you can, splitting 90 minutes into two 45-minute sessions is even better. You can also experiment with two strategies for better instructor engagement: 1) conduct a “pop quiz” after a break to make sure the audience understood the information they were just taught, and 2) don’t be afraid to adjust the breaks slightly to coincide with a change in topic. Changing topics at a break allows you to repeat the most important points three times. The first time when you introduced a topic, the second time when you have concluding remarks at the end of a topic, and a third time after the break when you make sure the audience understood the material you presented before the break.
What kind of surprise will engage your audience?
There are two strategies for using a surprise: 1) promise to surprise them in the future, or 2) don’t tell anyone until you surprise them. The first strategy works best when you are trying to get people to watch until the end of the training. This is commonly used by YouTubers to get people to watch the video until the end. Unfortunately, this backfires because we can fast-forward to the end. I’m not suggesting that you shouldn’t surprise your audience, but the surprise needs to delight your audience. You might also need to surprise and delight your audience more than once. Even then, some of your audience will still nod off and completely ignore you. When this happens, throw a Snickers bar at the offending student.
This is an essential tool for any instructor. It functions as a tool to prod sleeping students awake, is small enough to cause minimal injury when thrown, serves as an emergency food supply, and is gluten-free.
How to force-feed students legal stimulants
If legal counsel recommends against using projectiles to encourage class participation, you might also consider one of my all-time genius ideas–consuming dangerously large quantities of caffeine. I was scheduled for a two-day course in Ottawa, but the day before I needed to perform an audit in Pennsylvania. My flight was the last flight into Ottawa, which arrived at approximately 1 o’clock in the morning. My arrival was delayed an additional hour in customs by the person in front of me who was trying to smuggle an extra carton of smokes into the country. Just before 4 a.m., my taxi arrived at the Albert at Bay Suite Hotel. The class started at eight in the morning. I made it to class on time, and the excessive consumption of several pots of black coffee helped get me to lunch. Then my legs started getting a little shaky. Fortunately, there was a convenience store next door that sold my favorite chocolate–the Dark Aero bar! After four of these monstrous doses of cacao, and another pot of coffee, I could have listened to the lecture on the Canadian Medical Devices Regulations all night. The only problem is that my hands are still shaking 15 years later.
Hershey’s copied them, but the result was a mere shadow of Nestle’s greatness. Canadians know how to make junk food, tell a joke, and play hockey!
Why was the instructor engagement high in Ottawa?
Despite the physical handicap of sleep deprivation, I still learned a ton from my course in Canada. Here’s why:
The instructors were both regulatory experts that were able to share anecdotes, case studies, and stories about real-world application of the Canadian Medical Devices Regulations. One of the instructors even worked for Health Canada.
The audience was hyper-motivated to pass the course, because everyone in the class worked for a Notified Body that had sponsored them to take the course. In order to stay employed and get a raise, I needed to pass that course. If I failed the exam, I had to absorb the cost to travel back to Ottawa and retake the course in February (BRRRR!).
The instructors brought more than a dozen medical devices to the class. These props gave us something to read, touch, and ask questions about. The instructors broke us up into small teams to study the labeling and instructions for use of each device. Even students from Japan, Europe, and Australia were familiar with some of the products. This was critical because we all needed to be able to identify incorrect Canadian labeling.
The best instructor engagement tool used was humor. The instructor from Health Canada was hilarious. He had everyone laughing at his jokes for the entire course. Most of the jokes were not funny enough for a stand-up routine, but this was a mandatory regulatory course on Canadian regulations. Who would even expect a chuckle? Despite the strengths of these instructors, there is only one reason why I know the Canadian Medical Devices Regulations (CMDR), as well as I do. I use them every single week.
When students are forced to do homework, they will pay attention
After completing my CMDR training, I had to audit 162 days for BSI in 2011. Ninety percent of those 162 days were for companies that required a Canadian Medical Device License. This forced me to use the information I learned in the course. I was also consulting for companies at the same time I was auditing for BSI. Consulting clients hired me to prepare and submit the Canadian Medical Device License Applications for them. I also had to create procedures for Canadian Licensing, Incident Reporting, and Recalls. I spent another 60+ days in 2011 doing consulting, which helped me hone my knowledge of Canadian device regulations.
Teaching others will make you a guru
Most people are terrified to speak on any topic–even to a small group of coworkers. However, I believe that teaching others is the secret to becoming a guru on any topic. I was one of BSI’s instructors that taught the regulatory comparison course from 2010 to 2012, which compared the regulations of the USA, Canada, Europe, Australia, and Japan. Therefore, at least once a month, I had a classroom of 6-20 people asking me challenging questions about how to interpret and apply regulations from each of these countries to their products. I used every bit of knowledge I learned in that course in Ottawa, and I started using that knowledge immediately after the course. Peers, clients, and students challenge my knowledge of these topics every day. This is what makes you a subject matter expert. If you need to learn something about quality assurance or regulatory affairs watching a one-hour webinar, reading a blog, taking a five-day course, or shadowing another more experienced person is not enough. In the end, all of the above will get you to the level of barely competent! If you want to master any topic, you need to practice instructor engagement and use everything you learn for several years.
Learn how to create a regulatory plan for combining 510k with CE marking submissions in parallel instead of doubling your workload.
My first medical device regulatory submission was for CE Marking, while my second regulatory submission was for a 510k submission of the same product. Preparing submissions for different countries in parallel is a common path for medical device regulatory submissions, but it is also an inefficient path. If you know that you will be submitting both types of documents, then you should plan for this from the start and reduce your workload by at least 35%.
The reason why you can quickly reduce your workload by more than 65% is that both submission have very similar sections. Therefore, you can write the content for those sections in such a way that the material can be used for your 510k submission and CE Marking.
Identify duplicate sections in when combining 510k with CE marking projects
Most of your testing requirements should be identical when you are combining 510k with CE Marking submissions. However, the way the testing is presented is different. For your 510k submission you will attach the full testing report and write a brief statement about how the testing supports substantial equivalence. In contrast, CE marking technical files require a summary technical document or STED. The STED is a summary of each test that was performed. If you aren’t sure what testing is required, we created a test plan webinar to address this question specifically. Most of the work will be duplicated between your two test plans, but any outliers should be identified. For example, biocompatibility will need to include a biological evaluation plan (BEP) and biological evaluation report (BER), but this is optional for a 510k submission. There are also FDA requirements that are not required for CE marking, such as material mediated pyrogenicity testing and bacterial endotoxin testing for each production lot. In general, the possible testing categories are:
How to organize your medical device files when combining 510k with CE marking
The new FDA eSTAR has a unique PDF template that must be used for organizing your submission but Patrick Axtell, the person that helped create the FDA eSTAR templates, is also the Coordinator for the IMDRF Regulated Product Submission Working Group. He has inserted links in the eSTAR sections that cross-reference to the Regulated Product Submission Table of Content (i.e., RPS ToC). Therefore, best practice is to organize your medical device file in accordance with the RPS ToC:
Combining 510k with CE marking – how to construct your regulatory plan
In one of my previous blogs, I explained how the new FDA eSTAR template as a project management tool to verify that all of the section of a 510k submission are complete. Unfortunately, there is no CE Marking equivalent, but you can use your TF/MDR Index as a project management tool when you are constructing a combined plan for a 510k submission and CE Marking. The first step is to create a Index based on a recognized standard (EN standard or ISO standards). Historically we used the GHTF guidance document released by study group 1: N011:2008. When the EU MDR came into force, we added cross-references to the EU MDR in our TF/MDR Index. The GHTF guidance mirrors the format required in Annex III of the new EU MDR. I do not recommend using the NB-MED 2.5.1/rec 5 guidance document. Even though the content is similar to the GHTF guidance, the format is quite different. There is also a new IMDRF guidance document for Essential Principles of Safety and Performance that you should consider referencing.
Project and task management for your combined regulatory plan
If you are going to outsource sections of either submission, the sections should be written and reviewed by someone familiar with both types of submissions. The headers and footers will be unique to the type of submission, but I write the text in Google Docs without formatting for ease of sharing and so I can use my Chromebook.
If you have an in-house team that prepares your 510k submissions and Technical Files, you might consider training the people responsible for each section on the requirements for each type of submission. This eliminates rewriting and reformatting later. I like to assign who is writing each section in a separate column of my project management software. Then I will sort the sections by the expected date of completion. All the safety and performance testing, and any sections requiring validation, will typically be finished at the end of the project. Therefore, it is important to dedicate unique resources to those sections rather than asking one person to write several of those sections. You also will want to make sure any supporting documentation they need is completed early so that the project’s critical path doesn’t change.
Additional training for combining 510k with CE marking
We provide an on-demand 510k course series consisting of 33 FDA eSTAR webinars that you can purchase as a bundle or individually. We also have various training webinars about CE marking on our webinars page.
This article reviews the top reasons why other companies feel requesting FDA pre-sub meetings is a waste of time but you can’t afford to.
It only takes our consulting team a few hours to prepare an FDA pre-sub request using the new FDA PreSTAR. The FDA does not charge you a cent for submitting an FDA pre-sub, and a pre-sub should be part of every design plan. However, most companies are resistant to requesting FDA pre-sub meetings. In this article we review the top four reasons why companies resist submitting an FDA pre-sub request, and you will learn about the three options for the method of FDA feedback.
Our design is not finalized yet
The most common reason why people delay their request for an FDA pre-sub is that they are waiting until the design is complete. This rationale is flawed because the FDA can’t review data and the FDA can’t give you advice on the design of your device. The purpose of an FDA pre-sub is to “forge a better test plan.” The FDA prefers that you submit draft test protocols with specific questions. The ideal time to submit an FDA pre-sub request is 6-9 months before you approve your design outputs (i.e., design freeze). This timing should be shortly after you approve your design inputs (i.e., essential requirements for safety and performance, standards, and stakeholder requirements). Another reason for requesting a pre-sub early is that most companies need to submit a pre-sub supplement with a revised test plan. This is especially true for biocompatibility testing, non-clinical, benchtop performance testing plans, pre-clinical animal testing, human clinical studies, and human factors testing. The revised test plan includes protocol revisions based on the feedback from the original pre-sub.
It’s too late to request FDA pre-sub meetings
If you are less than a week away from submitting to the FDA, it is too late. The FDA target for scheduling an FDA pre-sub meeting is 70-75 days from the date your request was submitted. That’s 10-11 weeks. Most companies tell me that they plan to submit to the FDA within weeks or a couple of months, but most of the companies take nine months or longer. For example, what if your device fails EMC testing, and you have to change the design and retest for both EMC and electrical safety at an NRTL? At best, you will have an 8-week delay. If you submit a request next week, and everything goes as planned, you can always withdraw your request for the pre-sub. If you encounter a delay for any reason, suddenly, it’s not too late.
We don’t want to be bound by what the FDA says in the FDA pre-sub meeting
FDA pre-sub meetings are “non-binding.” That means that the FDA can change its mind, but it also means you don’t have to do everything the FDA says in an FDA pre-sub meeting. If you don’t ask a question about testing requirements, that doesn’t mean that the FDA does not have any testing requirements. The FDA knows what previous companies have submitted for testing better than you do, and they may be in the process of evaluating draft guidance documents. If you ask questions, you will have better insights into what the FDA expects. The most important questions are related to your rationale for why a specific testing specimen represents the “worst-case” for one of your tests. Selecting the wrong testing specimen will result in you repeating that test. Understanding FDA expectations helps you write better rationales for testing or test avoidance. You also might learn about deadlines for the implementation of new testing requirements that you might be able to avoid. Finally, you can ask the FDA about possible testing options you are considering if the FDA denies your most optimistic testing plans.
There is already a guidance document for our device
Not all device classifications have a guidance document explaining what information should be submitted in an FDA pre-market submission. However, there are almost one hundred Special Controls Guidance Documents, and for new device regulations (i.e., De Novo applications) the FDA now incorporates the special controls directly in the new regulation. Therefore, there is a good chance that the FDA published special controls as part of the regulation for your device or as a guidance document. As part of the special controls, the FDA defines what performance testing is required for your device. If you already know what testing is required, then the value in requesting FDA pre-sub meetings is diminished. But at least three other key benefits remain.
First, you can verify that the predicate you plan to use for comparative testing is not going to be a problem. Although the FDA can’t tell you which predicate to pick, the FDA can tell you if there is a problem with the predicate you have selected. This is especially important if the product is not currently registered and listed, because you may not know if the device was withdrawn from the market after it was cleared.
Second, not all testing standards are prescriptive. Many tests have testing options that require a decision. Input from the FDA may be valuable in making choices between various performance testing options. Sometimes, you forgo testing and provide a rationale instead. FDA feedback on any rationale for not doing testing is critical to prevent delays and requests for additional information later.
Third, there are many different FDA representatives who participate in FDA pre-sub meetings. The lead reviewer will invite specialists and the branch chief to the meeting. Each of these specialists can answer questions during a pre-submission meeting that they are not able to answer during the actual review process. You also have the opportunity to get feedback from the branch chief–who has insight from all the previous devices that were cleared with your product classification. Your lead reviewer is not likely to be as experienced as the branch chief, and may only have been working at the FDA for months. Your request for the 510k pre-sub meeting will help an inexperienced lead reviewer as much as it will help your company.
Which method of FDA pre-sub feedback should you request?
The FDA offers three different options for the method of feedback in pre-sub request:
a face-to-face meeting
a conference call
an email response
Feedback Option 1 – A Face-to-Face Meeting
Some executives believe that face-to-face meetings are critical in establishing relationships with people. However, you need to understand the culture of the people you are trying to build a relationship with. The FDA is an overworked bureaucracy, and government agencies have security concerns. When the FDA meets with visitors they must go to a different building and arrange for their guests to pass through security. This is more work and takes more time. To justify the extra work and time, you need a compelling reason why a face-to-face meeting with the FDA is necessary.
Traveling to the FDA will cost your team money and time that conference calls and emails will not. More importantly, you are limited to one hour for a pre-submission meeting. One hour is barely enough time to ask questions and listen to the answers. You only have minutes to introduce your company and your team and describe the product. There is no time for relationship building. The best way to impress the FDA is to 1) prepare thoroughly, 2) conduct an efficient meeting, and 3) ask smart questions.
There is one time when you should visit the FDA face-to-face–if you have a powerful demonstration and video just isn’t good enough.
Feedback Option 2 – Conference Call
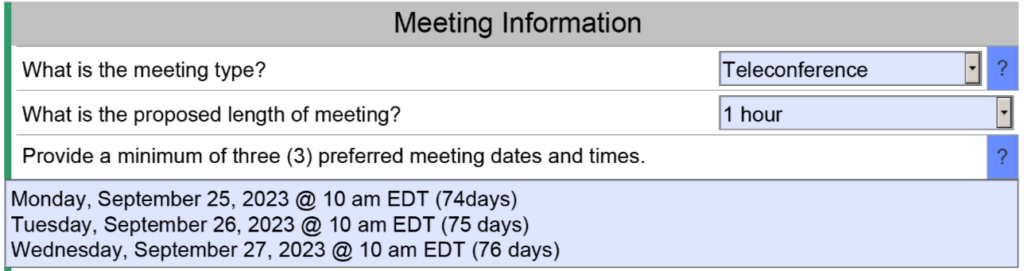
Conference calls save you time and money, but conference calls also save the FDA time and effort. You won’t personally meet people from the agency, but you can communicate information prior to the meeting and you can provide videos of simulated use for your device. Conference calls do have the advantage of allowing you to mute the call for a moment and make a comment among your team members without the agency listening as well. Whenever you are discussing a performance testing plan or a clinical study protocol with the FDA, you will probably want a conference call to enable clarification questions. The image below is an example of a request for a teleconference as the method of feedback. The FDA will still send an email response to your questions within 70 days, and you will want to submit your presentation slide deck for the teleconference at least 48 hours before the teleconference.
Feedback option 3 – Email
Email responses from the FDA are highly underrated in value. When you specify an email response, you generally receive a response to your questions sooner. You also should receive more information, because each person from the agency is able to provide an hour of their time to write detailed feedback. In a conference call, you are speaking for part of the hour, and only one person from the FDA can speak at a time. Therefore, you almost always have less feedback during conference calls and face-to-face meetings. The primary downside to email as a feedback method is that it is not interactive. In the case of submission issue review (SIR) requests (i.e., a special type of FDA pre-sub request), the FDA will only give you the option of a teleconference or email feedback–not both. Due to the challenges of scheduling teleconference, sometimes an email response can be delivered sooner and might be your best choice for an SIR request.
Learning More about FDA Pre-sub Meetings
If you would like to learn more about how to prepare the format and contents of an FDA pre-sub request, we have a four-part webinar series that will teach you how to prepare a pre-submission meeting request using the new FDA PreSTAR template.
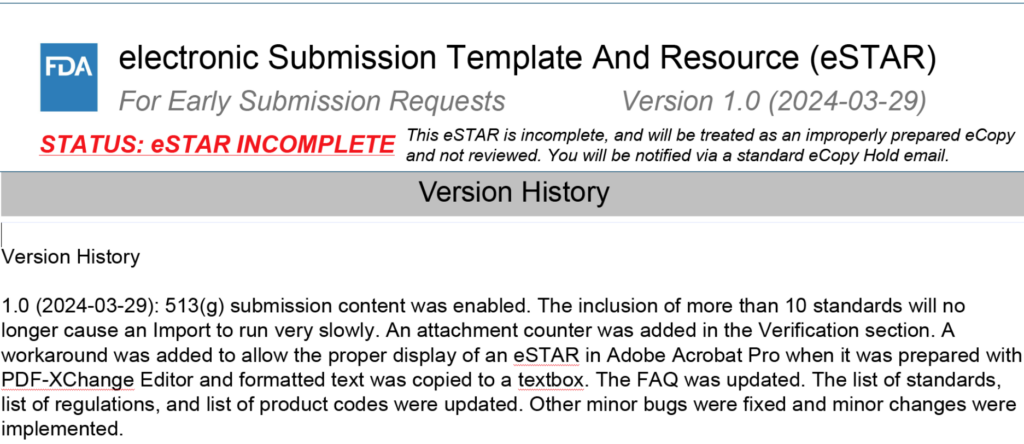
Version 1.0 of the FDA PreSTAR template, released March 29th, now enables the use of the PreSTAR template for 513g requests for information.
What is a 513g request?
A “513g” is a request for classification information from the FDA. The reference is to a Food, Drug & Cosmetic Act section. The purpose of the submission is to ask the FDA what product classification would be most appropriate for your device and what the appropriate regulatory pathway will be. The regulation requires the FDA to provide a written response within 60 days of receiving the 513g request. The submission also requires payment of an FDA user fee eligible for a small business discount.
Is it required to use the new FDA PreSTAR v1.0 template for a 513g request?
No, the FDA PreSTAR is not required to submit a 513g request for information. The FDA has not updated the 2019 guidance document yet, and the FDA continues to allow the use of an FDA eCopy for 513g submissions. However, the updated PreSTAR template simplifies the process of submitting a request for classification information.
What is the required content of a 513g request?
Page 15 of the FDA guidance for 513g requests specifies the following content:
cover letter,
description of the device,
description of what the device is to be used for,
any proposed labeling or promotional material for the device and, as applicable, any labeling or promotional material of a similar, legally marketed device, if available.
The guidance also details the minimum requirements for these four content requirements. The cover letter requirements specified in the guidance include “your specific question(s) concerning the class in which a device has been classified and/or the regulatory requirements applicable to a device.” When the PreSTAR is used for a pre-submission, there is a designated section at the end of the template for entering questions. However, v1.0 does not allow this option for a 513g. Therefore, questions must be added to the cover letter instead. The template Medical Device Academy created for a 513g includes the following default question:
Reason for the 513(g) Submission:
[Company Name] plans to submit a pre-market submission in 202x, and the company is requesting a decision from the FDA regarding the regulatory pathway for the subject device.
This section can be modified to include additional questions, depending on the specific reason for the 513g request.
When should a 513g request for information be submitted?
Usually, device companies ask me if I think they should submit a 513g or a pre-submission request to answer questions about the testing requirements. Often, the device has a known product classification code that requires a submission of 510(k). Sometimes, there will even be a Special Controls Guidance document available for the product classification. In these situations, a 513g is entirely unnecessary. I can understand the difficulties people experience when navigating the FDA product classification database because the database does not use modern natural language search algorithms like Google. However, a greater concern is that most companies ask this question after they have already started the development of their device and before they plan to initiate design verification testing. This is very late in the design process, and it is even a little late to conduct a pre-submission request. Your 513g submission should be during the beginning of your design project (i.e., during the concept or feasibility phases of design) to verify the proposed regulatory pathway.
How to prepare a 513g
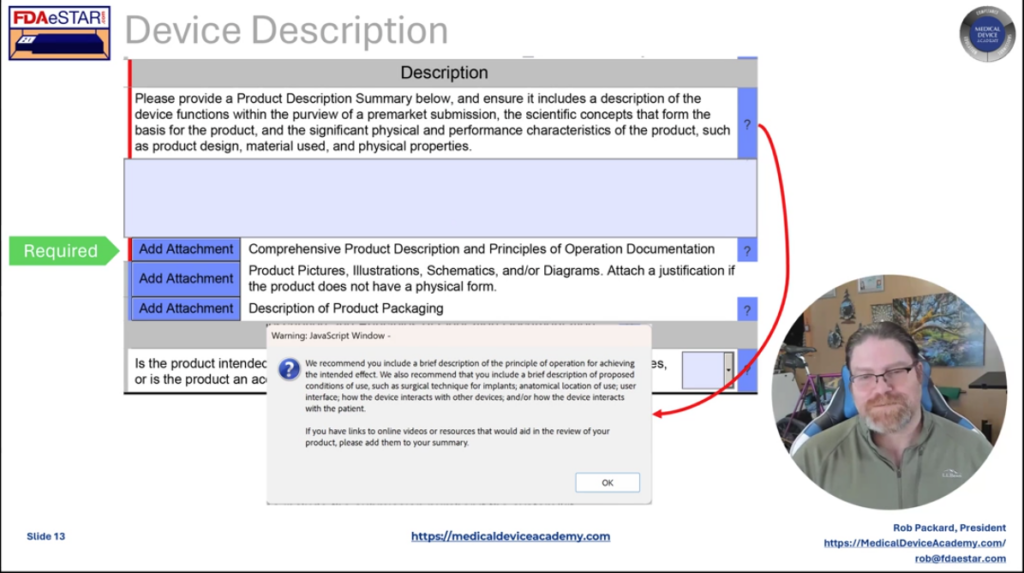
For any device submission, including a 513g, you must prepare a detailed device description for the FDA. Many companies find this difficult. Therefore, we provide a template for the device description. In addition to the device description, we recommend including a copy of the draft labeling and instructions for use (IFU) with each device submission. A pre-submission does not require draft labeling, but a 513g classification request does to ensure the FDA understands your intended use for the device. Therefore, we provide templates for companies to prepare these drafts.
Other Resources
If you need to submit a 513g request, you can learn more about FDA content requirements by watching our 513g submission webinar. You will also receive access to our 513g templates if you purchase the webinar bundle. We also provide the templates for the device description, draft label, and draft instructions for use (IFU) to new clients submitting a pre-submission meeting request, a 510k submission, or a De Novo Classification Request. In addition, there are six (6) other templates included with the 513g webinar bundle. Those templates are specifically required for De Novo submissions, and we recommend including them if you believe your device requires a De Novo submission.
Using the new FDA eSTAR template also requires a new process for eSTAR project management to prepare your 510k and De Novo submissions.
Outline of ten (10) major changes resulting from the new FDA eSTAR template
As of October 1, 2023, all 510k and De Novo submissions to the FDA now require using the new FDA eSTAR template and the template must be uploaded to the FDA Customer Collaboration Portal (CCP). Yesterday the FDA published an updated guidance explaining the 510k electronic submission requirements, but there are ten (10) major changes to Medical Device Academy’s submission process resulting from the new eSTAR templates:
We no longer need a table of contents.
We no longer use the volume and document structure.
We are no longer required to conform to sectioning or pagination of the entire submission.
We no longer worry about the RTA screening or checklist (it doesn’t exist).
We no longer bother creating an executive summary (it’s optional).
We no longer have a section for Class 3 devices, because there are no Class 3 510(k) devices anymore.
We no longer use FDA Form 3514, because that content is now incorporated into the eSTAR.
We no longer create a Declaration of Conformity, because the eSTAR creates one automatically.
We no longer recommend creating a 510(k) Summary, because the eSTAR creates one automatically
We no longer use FedEx, because we can upload to FDA CCP electronically instead.
What is different in the 510k requirements?
Despite all the perceived changes to the FDA’s pre-market notification process (i.e., the 510k process), the format and content requirements have not changed much. The most significant recent change to the 510k process was the requirement to include cybersecurity testing.
Outline of eSTAR Project Management
There were 20 sections in a 510k submission. Medical Device Academy’s consulting team created a template for the documents to be included in each section. eSTAR project management is different because there are no section numbers to reference. To keep things clear, we recommend using one or two words at the beginning of each file name to define the section it belongs in. The words should match up with the bookmarks used by the FDA. However, you should be careful not to make the file names too long. Below is a list of all of the sections:
The Benefit, Risks, and Mitigation Measures Section only applies to De Novo Classification Requests. The Quality Management Section includes subsections for Quality Management System Information, Facility Information, Post-Market Studies, and References. However, only the References subsection will be visible in most submissions because the other three subsections are part of the Health Canada eSTAR pilot. Other sections and subsections will be abbreviated or hidden depending on the dropdown menu selections you select in the eSTAR. For example, the cybersecurity section will remain hidden if your device does not have wireless functionality or a removable storage drive.
A Table of Contents is no longer required for 510k submissions
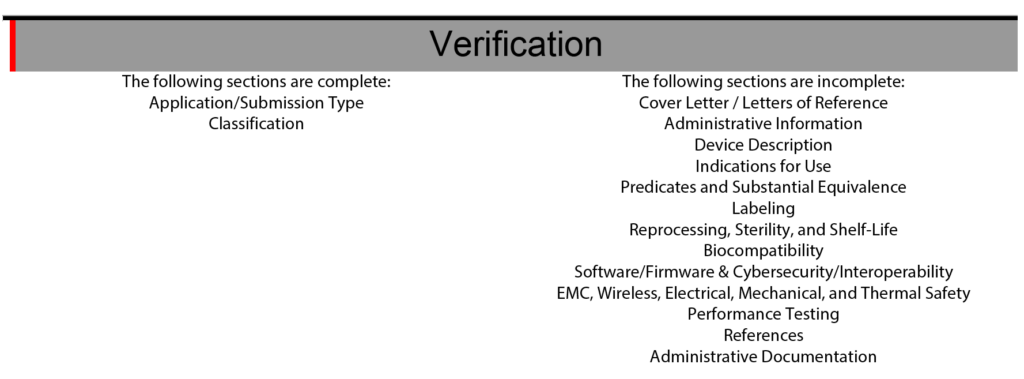
510k submissions using the FDA eCopy format required a Table of Contents, and Medical Device Academy used the Table of Contents as a project management tool. Sometimes, we still use our Table of Contents template to communicate assignments and manage the 510k project. The sections of the Table of Contents would also be color-coded green, blue, yellow, and red to communicate the status of each section. FDA eSTAR project management uses a similar color coding process with colored bars on the side of the template to indicate if the section is incomplete, complete, or optional.
The eSTAR also has a verification section at the end of the template to help with eSTAR project management. The verification section lists each of the 13 major sections of an FDA eSTAR. When the sections are completed, the section’s name automatically moves from the right side of the verification section to the left side. During the past two years (2021 – 2023) of implementing the eSTAR template, I have slowly learned to rely only on the eSTAR to communicate the status of each section. To assign responsibilities for each section of the 510k submission, we still use the Table of Contents simple lists and project management tools like Monday.com. Using the eSTAR verification section to check on the status of each 510k section also increases our team’s proficiency with the eSTAR every time we use it.
Using Dropbox for eSTAR project management
PreSTAR templates for a Q-Sub meeting are approximately half the length (i.e., 15 pages instead of 30+ pages) of an eSTAR template, and the 510k submission requires far more attachments than a Q-Sub. Therefore, we can usually email a revised draft of the PreSTAR to a team member for review, but we can’t use email to share a nearly complete eSTAR with a team member. Therefore, Medical Device Academy uses Dropbox to share revisions of the eSTAR between team members. Some of our clients use One Drive or Google Drive to share revisions. We also create sub-folders for each type of testing. This keeps all of the documents and test reports for a section of the eSTAR in one place. For example, the software validation documentation will be organized in one sub-folder of the Dropbox folder for a 510k project.
When using FDA eCopies instead of the FDA eSTAR template, we used twenty subfolders labeled and organized by volume numbers 1-20. Some of those 20 sections are now obsolete (e.g., Class III Summary), and others (e.g., Indications for Use) are integrated directly into the eSTAR template. Therefore, a team may only need 8-10 sub-folders to organize the documents and test reports for a 510k project. We typically do not attach these documents and test reports until the very end of the submission preparation because if the FDA releases a new version of the eSTAR, the attachments will not export from an older version of the eSTAR to the new version.
Coordination of team collaboration is critical to successful eSTAR project management
In the past, Medical Device Academy always used a volume and document structure to organize an FDA eCopy because this facilitated multiple team members simultaneously working on the same 510k submission–even from different countries. Many clients will use SharePoint or Google Docs to facilitate simultaneous collaboration by multiple users. Unfortunately, the eSTAR cannot be edited by two users simultaneously because it is a secure template that can only be edited in Adobe Acrobat Pro. Therefore, the team must communicate when the eSTAR template is being updated and track revisions. For communication, we use a combination of instant messenger apps (e.g., Slack or Whatsapp) and email, while revisions are tracked by adding the initials and date of the editor to the file name (e.g., nIVD 4.3 rvp 12-5-2023.pdf).
Importance of peer reviews
Each section of the FDA eSTAR must be completed before the submission can be uploaded to the Customer Collaboration Portal (CCP). If the FDA eSTAR is incomplete, the CCP will identify the file as incomplete. You will not be able to upload the file. If questions in the eSTAR are incorrectly answered, then sections that should be completed may not be activated because of how the questions were answered. Below are two examples of how the eSTAR questions can be incorrectly answered.
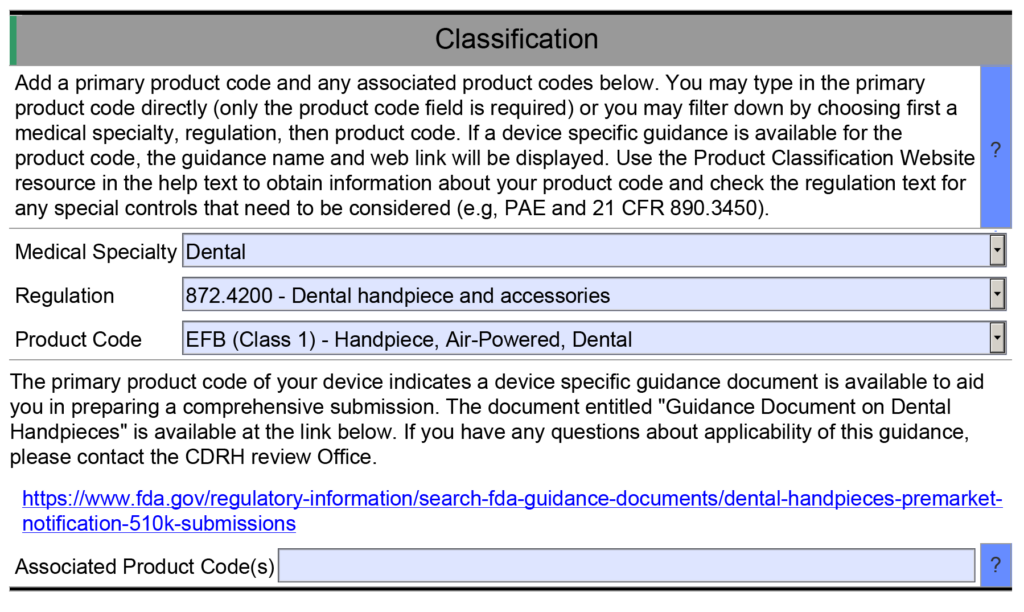
Example 1 – One of the helpful resource features of the FDA eSTAR is that many fields are populated with a dropdown menu of answers. One example is found in the Classification section of the eSTAR. This section requires the submitter to identify the device’s classification by answering three questions: 1) review panel, 2) classification regulation, and 3) the three-letter product code. Each of these fields uses a dropdown menu to populate the field, and the dropdown options for questions two and three depend on answers to the previous question. However, if you manually type the product code into the field for the third question, then the eSTAR will not identify any applicable special controls guidance documents for your device. Unless you are already aware of an applicable special controls guidance document, you will answer questions in the eSTAR about special controls with “N/A.” The eSTAR will only identify a special controls guidance document for your device if you select a product code from the dropdown menu, but the FDA reviewer knows which special controls guidance documents are applicable. This is why the FDA performs a technical screening of the eSTAR before the substantive review begins.
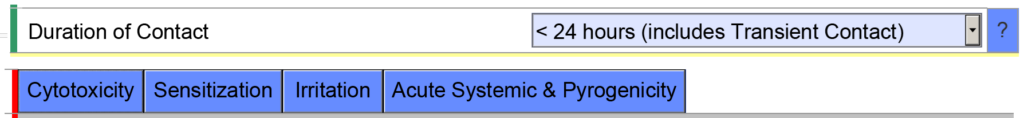
Example 2 – If you indicate the cumulative duration of contact for an externally communicating device < 24 hours, the eSTAR template will expect you to evaluate the following biocompatibility endpoints: cytotoxicity, sensitization, irritation, systemic toxicity, and pyrogenicity.
However, if you indicate the cumulative duration of contact is < 30 days, the eSTAR template will be populated with additional biocompatibility endpoints. The eSTAR doesn’t know what the cumulative duration of use is, but the FDA reviewer will. This is why the FDA performs a technical screening of the eSTAR before the substantive review begins.
To make sure that all of the sections of your submission are complete, it’s helpful to have a second person review all of the answers to make sure that everything was completed correctly. Even experienced consultants who prepare 510k submissions every week can make a mistake and incorrectly answer a question in one of the eSTAR fields. Therefore, you shouldn’t skip this critical QC check.
This is an overview of the updated 510k electronic submission guidance document that the FDA released on October 2, 2023.
What’s included in the 510k electronic submission guidance?
As with any FDA guidance, there is an introduction and background regarding the reason for the updated guidance document (i.e., eSTAR guidance). At the very beginning of the document (i.e., page 3) the reference to the RTA Guidance was deleted, because there is no longer an RTA screening process with the implementation of the FDA eSTAR templates. The updated guidance explains on page 6 that “The CDRH Portal will automatically verify that the eSTAR is complete, and therefore we do not expect to receive incomplete 510(k) eSTARs.” In the scope section, the FDA specifies that this document is specific to 510k submissions using the eSTAR template. The document also explains that CBER conducted a pilot with the eSTAR template in June 2022 and now the FDA eSTAR template must be used in conjunction with the CDRH Portal for submission of a 510k to CBER. The FDA has plans to release a similar De Novo submission guidance for using the eSTAR template, but this has not happened in the year since the FDA announced the intention to do so. In the “Significant Terminology” section of the guidance (i.e., Section IV), the FDA provides definitions for each of the different types of submissions: eCopy, eSubmitter, etc. In the “Current Electronic Submission Template Structure, Format, and Use” section of the guidance (i.e., Section V), the FDA modified the term used for the company that is applying for 510k clearance from “Submitter” to “Applicant,” because sometimes a regulatory consultant or 3rd party reviewer is submitting the 510k on behalf of the applicant. On page 12 of the updated guidance, the FDA added “Withdrawal requests” to the list of 510k submissions/information that is exempt from the 510k electronic submission requirements. In the next to last section of the electronic submission guidance, the FDA provides a table outlining all of the sections of the new eSTAR template. The table is reproduced later in this article. If you are interested in a tutorial on completing each section outlined in the table, we recommend purchasing Medical Device Academy’s 510(k) Course. The last section of the eSTAR guidance indicates the timing for compliance with the updated guidance (i.e., October 1, 2023).
What is the deadline for compliance with the guidance?
The deadline has now passed. The new eSTAR template must be used for all 510k and De Novo submissions as of October 1, 2023. You must upload the new FDA eSTAR submissions using the CDRH Portal. You will need to request an account using a registration hyperlink.
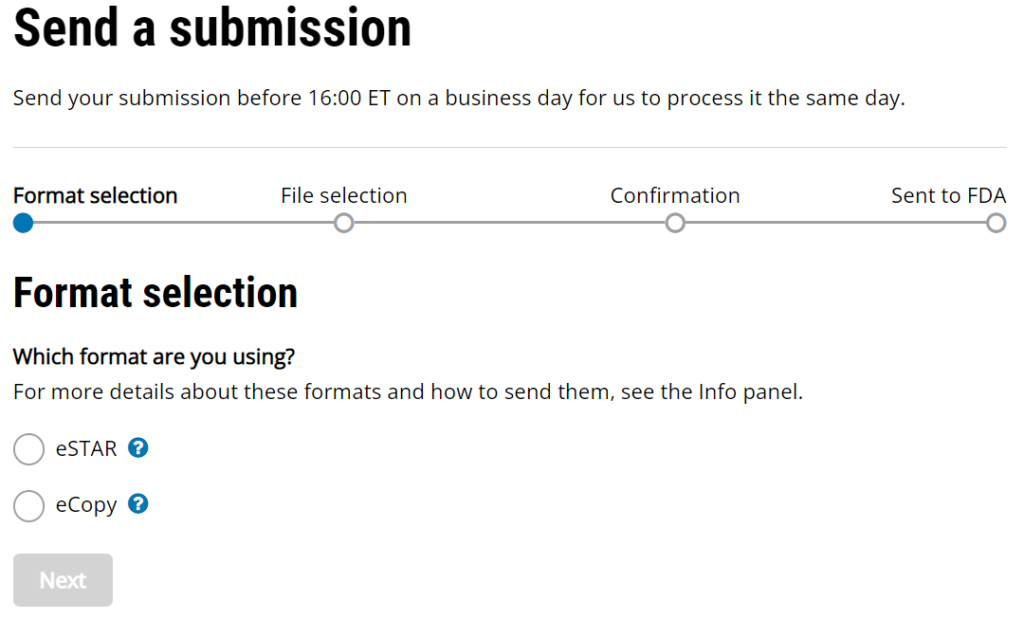
What’s missing from this 510k submission guidance?
The updated 510k electronic submission guidance does not provide information regarding the receipt date for electronic submissions made through the new customer collaboration portal (CCP) created by CDRH. The image below is a screen capture of the current CCP upload webpage. It includes the following statement, “Send your submission before 16:00 ET on a business day for us to process it the same day.” This statement was added sometime in August or September, but the FDA has not released a detailed explanation. This statement makes it clear that the FDA is not promising to process a submission the “same day” if the submission is received after 4:00 p.m. ET. However, “processed” does not have the same meaning as “receipt date.”
Another element missing from this updated guidance is a reference to human factors documentation. For any devices that have a user interface that is different from the predicate device, and for software devices, the FDA requires documentation of your human factors process to make sure that differences in the user interface do not result in new or different risks when compared to the predicate device. The 2016 FDA guidance for human factors has not been updated, but FDA reviewers continue to issue deficiencies related to the objective evidence provided in a 510k for human factors validation.
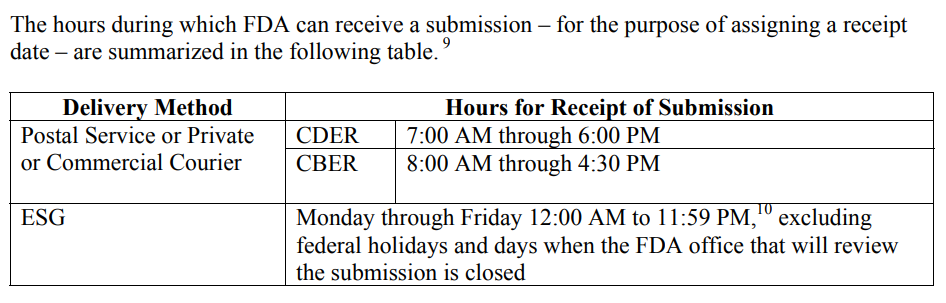
The FDA must be consistent in the wording for “Hours for Receipt of Submission” because this affects submissions at the end of the fiscal year, but it also affects any submissions with a deadline for response to an RTA Hold, AI Response, and IDE submissions. The CDER and CBER divisions of the FDA address the need for defining the date of receipt in a guidance document specific to this topic, “Providing Regulatory Submissions in Electronic Format–Receipt Date.” Below is a screen capture copied from page 4 of the guidance.
Another element missing from this new guidance is a reference to human factors documentation. For any devices that have a user interface that is different from the predicate device, and for software devices, the FDA requires documentation of your human factors process to make sure that differences in the user interface do not result in new or different risks when compared to the predicate device. The 2016 FDA guidance for human factors has not been updated, but FDA reviewers continue to issue deficiencies related to the objective evidence provided in a 510k for human factors validation.
What are the new sections for a 510k submission?
In 2019, the FDA released a guidance document on the “Format of Traditional and Abbreviated 510(k)s.” That guidance outlines the 20 sections of a traditional 510k submission that have been used for decades. However, the new 510k electronic submission guidance has no numbering for the sections of the eSTAR template, and there are 22 sections instead of 20 sections. Several of the new sections are elements of the current FDA submission cover sheet (i.e., FDA Form 3514), and some sections exist in the 2019 guidance that were eliminated, such as: “Class III Summary and Certification.” Therefore, Medical Device Academy is recreating 100% of our 510k training webinars to explain how our 510k templates are used with the 510k eSTAR template and how to fill in the PDF form. To prevent confusion between the two formats, we are using letters for each section in the eSTAR template instead of numbers (i.e., A-V instead of 1-20). Table 1 from the new eSTAR guidance is reproduced below for your information.
Information Requested
Description
A
Submission Type
Identification of key information that may be useful to FDA in the initial processing and review of the 510(k) submission, including content from current Form FDA 3514, Section A.
B
Cover Letter / Letters of Reference
Attach a cover letter and any documents that refer to other submissions.
C
Submitter Information
Information on submitter and correspondent, if applicable, consistent with content from current Form FDA 3514, Sections B and C.
Information on prior submissions for the same device included in the current submission, such as submission numbers for a prior not substantially equivalent (NSE) determination, prior deleted or withdrawn 510(k), Q-Submission, Investigational Device Exemption (IDE) application, premarket approval (PMA) application, humanitarian device exemption (HDE) application, or De Novo classification request.
E
Consensus Standards
Identification of voluntary consensus standard(s) used, if applicable. This includes both FDA-recognized and nonrecognized consensus standards.
F
Device Description
Identification of listing number if listed with FDA.Descriptive information for the device, including a description of the technological characteristics of the device including materials, design, energy source, and other device features, as defined in section 513(i)(1)(B) of the FD&C Act and 21 CFR 807.100(b)(2)(ii)(A). Descriptive information also includes a description of the principle of operation for achieving the intended effect and the proposed conditions of use, such as surgical technique for implants; anatomical location of use; user interface; how the device interacts with other devices; and/or how the device interacts with the patient.Information on whether the device is intended to be marketed with accessories.Identification of any applicable device-specific guidance document(s) or special controls for the device type as provided in a special controls document (or alternative measures identified that provide at least an equivalent assurance of safety and effectiveness) or in a device-specific classification regulation, and/or performance standards. See “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].”
G
Proposed Indications for Use (Form FDA 3881)
Identification of the proposed indications for use of the device. The term indications for use, as defined in 21 CFR 814.20(b)(3)(i), describes the disease or condition the device will diagnose, treat, prevent, cure, or mitigate, including a description of the patient population for which the device is intended.
H
Classification
Identification of the classification regulation number that seems most appropriate for the subject device, as applicable.
I
Predicates and Substantial Equivalence
Identification of a predicate device (e.g., 510(k) number, De Novo number, reclassified PMA number, classification regulation reference, if exempt and limitations to exemption are exceeded, or statement that the predicate is a preamendments device).The submission should include a comparison of the predicate and subject device and a discussion why any differences between the subject and predicate do not impact safety and effectiveness [see section 513(i)(1)(A) of the FD&C Act and 21 CFR 807.87(f)]. A reference device should also be included in the discussion, if applicable. See “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].”
J
Design/Special Controls, Risks to Health, and Mitigation Measures
Applicable to Special 510(k) submissions only.Identification of the device changes and the risk analysis method(s) used to assess the impact of the change(s) on the device and the results of the analysis.Risk control measures to mitigate identified risks (e.g., labeling, verification). See “The Special 510(k) Program.”
K
Labeling
Submission of proposed labeling in sufficient detail to satisfy the requirements of 21 CFR 807.87(e). Generally, if the device is an in vitro diagnostic device, the labeling must also satisfy the requirements of 21 CFR 809.10. Additionally, the term “labeling” generally includes the device label, instructions for use, and any patient labeling. See “Guidance on Medical Device Patient Labeling.”
Summary of methods used to establish that device performance is maintained for the entirety of the proposed shelf-life (e.g., mechanical properties, coating integrity, pH, osmolality), if applicable.
For non-in vitro diagnostic devices: Provide information on the non-clinical and clinical test reports submitted, referenced, or relied on in the 510(k) for a determination of substantial equivalence. See “Recommended Content and Format of NonClinical Bench Performance Testing Information in Premarket Submissions.”For in vitro diagnostic devices: Provide analytical performance, comparison studies, reference range/expected values, and clinical study information.
T
References
Inclusion of any literature references, if applicable.
U
Administrative Documentation
Inclusion of additional administrative forms applicable to the submission, including but not limited to a general summary of submission/executive summary (recommended), a Truthful and Accuracy Statement, and a 510(k) Summary or statement.
V
Amendment/Additional Information (AI) response
Inclusion of responses to Additional Information requests.
Important information in the eSTAR guidance
In Table 1 above, there are 14 hyperlinks to various FDA guidance documents. These links are extremely helpful when you have questions about a specific question. Unfortunately, the 510k electronic submission guidance document will quickly become out-of-date as guidance documents are updated and made obsolete. In particular, one of the A-list final guidance documents that was planned for FY 2023 was the FDA cybersecurity guidance. The updated cybersecurity guidance was finally released last week.
The FDA released a new draft 510k predicate selection guidance on September 7, but the draft guidance proposes controversial additions.
Download the Draft FDA Predicate Selection Guidance
On September 7, 2023, a draft predicate selection guidance document was released by the FDA. Normally the release of a new draft of FDA guidance documents is anticipated and there is an obvious need for the draft. This new draft, however, appears to include some controversial additions that I feel should be removed from the guidance. This specific guidance was developed to help submitters use best practices in selecting a predicate. There is some useful advice regarding the need to review the FDA database for evidence of use-related and design-related safety issues associated with a potential predicate that is being considered. Unfortunately, the last section of the guidance suggests some controversial recommendations that I strongly disagree with.
Please submit comments to the FDA regarding this draft guidance
This guidance is a draft. Your comments and feedback to the FDA will have an impact on FDA policy. We are preparing a redlined draft of the guidance with specific comments and recommended changes. We will make the comments and feedback available for download from our website on our predicate selection webinar page. We are also creating a download button for the original draft in Word (.docx) format, and sharing the FDA instructions for how to respond.
Section 1 – Introduction to the guidance
The FDA indicates that this new draft predicate selection guidance document was created to provide recommendations to implement four (4) best practices when selecting a predicate device to support a 510k submission. This first objective is something that our consulting firm recommended in a training webinar. The guidance indicates that the guidance was also created by the FDA in an attempt to improve the predictability, consistency, and transparency of the 510k pre-market review process. This second objective is not accomplished by the draft guidance and needs to be modified before the guidance is released as a final guidance.
Section 2 – Background
This section of the guidance is divided into two parts: A) The 510k Process, and B) 510k Modernization.
A. The 510k Process
The FDA released a Substantial Equivalence guidance document that explains how to demonstrate substantial equivalence. The guidance document includes a new decision tree that summarizes each of the six questions that 510k reviewers are required to answer in the process of evaluating your 510k submission for substantial equivalence. The evidence of substantial equivalence must be summarized in the Predicates and Substantial Equivalence section of the FDA eSTAR template in your 510k submission, and the guidance document reviews the content that should be provided.
Substantial equivalence is evaluated against a predicate device or multiple predicates. To be considered substantially equivalent, the subject device of your 510k submission must have the same intended use AND the same technological characteristics as the predicate device. Therefore, you cannot use two different predicates if one predicate has the same intended use (but different technological characteristics), and the second predicate has the same technological characteristics (but a different intended use). That’s called a “split predicate,” and that term is defined in the guidance This does not prohibit you from using a secondary predicate, but you must meet the requirements of this guidance document to receive 510k clearance. The guidance document reviews five examples of multiple predicates being used correctly to demonstrate substantial equivalence.
B. 510k Modernization
The second part of this section refers to the FDA’s Safety Action Plan issued in April 2018. The announcement of the Safety Action Plan is connected with the FDA’s announcement of actions to modernize the 510k process as well. The goals of the FDA Safety Action Plan consist of:
Establish a robust medical device patient safety net in the United States
Explore regulatory options to streamline and modernize timely implementation of postmarket mitigations
Spur innovation towards safer medical devices
Advance medical device cybersecurity
Integrate the Center for Devices and Radiological Health’s (CDRH’s) premarket and postmarket offices and activities to advance the use of a TPLC approach to device safety
Examples of modernization efforts include the following:
Conversion of the remaining Class 3 devices that were designated for the 510k clearance pathway to the PMA approval process instead
Use of objective performance standards when bringing new technology to the market
Use of more modern predicate devices (i.e., < 10 years old)
In this draft predicate selection guidance, the FDA states that feedback submitted to the docket in 2019 has persuaded the FDA to acknowledge that focusing only on modern predicate devices may not result in optimal safety and effectiveness. Therefore, the FDA is now proposing the approach of encouraging best practices in predicate selection. In addition, the draft guidance proposes increased transparency by identifying the characteristics of technological characteristics used to support a 510k submission.
The FDA did not mention an increased emphasis on risk analysis or risk management in the guidance, but the FDA is modernizing the quality system regulations (i.e., 21 CFR 820) to incorporate ISO 13485:2016 by reference. Since ISO 13485:2016 requires the application of a risk-based approach to all processes, the application of a risk-based approach will also impact the 510k process in multiple ways, such as design controls, supplier controls, process validation, post-market surveillance, and corrective actions.
Section 3 – Scope of the predicate selection guidance
The draft predicate selection guidance indicates that the scope of the guidance is to be used in conjunction with the FDA’s 510k program guidance. The scope is also not intended to change to applicable statutory or regulatory standards.
Section 4 – How to use the FDA’s predicate selection guidance
The FDA’s intended use of the predicate selection guidance is to provide submitters with a tool to help them during the predicate selection process. This guidance suggests a specific process for predicate selection. First, the submitter should identify all of the possible legally marketed devices that also have similar indications for use. Second, the submitter should exclude any devices with different technological characteristics if the differences raise new or different issues of risk. The remaining sub-group is referred to in the guidance as “valid predicate device(s).” The third, and final, step of the selection process is to use the four (4) best practices for predicate selection proposed in the guidance. The diagram below provides a visual depiction of the terminology introduced in this guidance.
Section 5 – Best Practices (for predicate selection)
The FDA predicate selection guidance has four (4) best practices recommended for submitters to use when narrowing their list of valid predicate devices to a final potential predicate(s). Prior to using these best practices, you need to create a list of legally marketed devices that could be potential predicates. The following FDA Databases are the most common sources for generating a list of legally marketed devices:
Our team usually uses the Basil Systems Regulatory Database to perform our searches. Basil Systems uses data downloaded directly from the FDA, but the software gives us four advantages over the FDA public databases:
The search engine uses a natural-language algorithm rather than a Boolean search.
The results include analytics regarding the review timelines and a “predicate tree.”
Basil Systems also has a post-market surveillance database that includes all of the FDA adverse events and recall data, but it also includes access to data from Health Canada and the Australian TGA.
A. Predicate devices cleared using well-established methods
Some 510k submissions use the same methods used by a predicate device that was used for their substantial equivalence comparison, while other devices use well-established methods. The reason for this may have been that the 510k submission preceded the release of an FDA product-specific, special controls guidance document. In other cases, the FDA may not have recognized an international standard for the device classification. You can search for recognized international standards associated with a specific device classification by using the FDA’s recognized consensus standards database. An example is provided below.
New 510k submissions should always use the methods identified in FDA guidance documents and refer to recognized international standards instead of copying the methods used to support older 510k submissions that predate the current FDA guidance or recognized standards. The problem with the FDA’s proposed approach is that the FDA is implying that a device that was not tested to the current FDA guidance or recognized standards is inherently not as safe or effective as another device that was tested to the current FDA guidance or recognized standards. This inference may not be true. Therefore, even though this may be a consideration, it is not appropriate to require manufacturers to include this as a predicate selection criterion. The FDA is already taking this into account by requiring companies to comply with the current FDA guidance and recognized standards for device description, labeling, non-clinical performance testing, and other performance testing. An example of how the FDA PreSTAR automatically notifies you of the appropriate FDA special controls guidance for a product classification is provided below.
B. Predicate devices meet or exceed expected safety and performance
This best practice identified in the FDA predicate selection guidance recommends that you search through three different FDA databases to identify any reported injury, deaths, or malfunctions of the predicate device. Those three databases are:
All of these databases are helpful, but there are also problems associated with each database. In general, adverse events are underreported, and a more thorough review of post-market surveillance review is needed to accurately assess the safety and performance of any device. The MAUDE data represents reports of adverse events involving medical devices and it is updated weekly. The data consists of all voluntary reports since June 1993, user facility reports since 1991, distributor reports since 1993, and manufacturer reports since August 1996. The MDR Data is no longer updated, but the MDR database allows you to search the CDRH database information on medical devices that may have malfunctioned or caused a death or serious injury during the years 1992 through 1996. Medical Product Safety Network (MedSun) is an adverse event reporting program launched in 2002 by CDRH. The primary goal for MedSun is to work collaboratively with the clinical community to identify, understand, and solve problems with the use of medical devices. The FDA predicate selection guidance, however, does not mention the Total Product Life Cycle (TPLC) database which is a more efficient way to search all of the FDA databases–including the recall database and the 510k database.
The biggest problem with this best practice as a basis for selecting a predicate is that the number of adverse events depends upon the number of devices used each year. For a small manufacturer, the number of adverse events will be very small because there are very few devices in use. For a larger manufacturer, the number of adverse events will be larger–even though it may represent less than 0.1% of sales. Finally, not all companies report adverse events when they are required to, while some companies may over-report adverse events. None of these possibilities is taken into consideration in the FDA’s draft predicate selection guidance.
C. Predicate devices without unmitigated use-related or design-related safety issues
For the third best practice, the FDA predicate selection guidance recommends that submitters search the Medical Device Safety database and CBER Safety & Availability (Biologics) database to identify any “emerging signals” that may indicate a new causal association between a device and an adverse event(s). As with all of the FDA database searches, this information is useful as an input to the design process, because it helps to identify known hazards associated with similar devices. However, a more thorough review of post-market surveillance review is needed to accurately assess the safety and performance of any device–including searching databases from other countries where similar devices are marketed.
D. Predicate devices without an associated design-related recall
For the fourth best practice, the FDA predicate selection guidance recommends that submitters search the FDA recalls database. As stated above, the TPLC database includes this information for each product classification. Of the four best practices recommended by the FDA, any predicate device that was to a design-related recall is unlikely to be accepted by the FDA as a suitable predicate device. Therefore, this search should be conducted during the design planning phase or while design inputs are being identified. If you are unable to identify another predicate device that was not the subject of a design-related recall, then you should request a pre-submission meeting with the FDA and provide a justification for the use of the predicate device that was recalled. Your justification will need to include an explanation of the risk controls that were implemented to prevent a similar malfunction or use error with your device. Often recalls result from quality problems associated with a supplier that did not make a product to specifications or some other non-conformity associated with the assembly, test, packaging, or labeling of a device. None of these problems should automatically exclude the use of a predicate because they are not specific to the design.
Section 6 – Improving Transparency
This section of the FDA predicate selection guidance contains the most controversial recommendations. The FDA is proposing that the 510k summary in 510k submissions should include a narrative explaining their selection of the predicate device(s) used to support the 510k clearance. This would be a new requirement for the completion of a 510k summary because that information is not currently included in 510k summaries. The new FDA eSTAR has the ability to automatically generate a 510k summary as part of the submission (see example below), but the 510k summary generated by the eSTAR does not include a section for including a narrative explaining the reasons for predicate selection.
The FDA added this section to the draft guidance with the goals of improving the predictability, consistency, and transparency of the 510k pre-market review process. However, the proposed addition of a narrative explaining the reasons for predicate selection is not the best way to achieve those goals. Transparency is best achieved by eliminating the option of a 510k statement (i.e., 21 CFR 807.93). Currently, the 510k process allows for submitters to provide a 510k statement or a 510k summary. The 510k statement prevents the public from gaining access to any of the information that would be provided in a 510k summary. Therefore, if the narrative explaining the reasons for predicate selection is going to be required in a 510k submission, that new requirement should be added to the substantial equivalence section of the eSTAR instead of only including it in the 510k summary. If the 510k statement is eliminated as an option for submitters, then all submitters will be required to provide a 510k summary and the explanation for the predicate selection can be copied from a text box in the substantial equivalence section.
The FDA eSTAR ensures consistency of the 510k submission contents and format, and tracking of FDA performance has improved the consistency of the FDA 510k review process. Adding an explanation for predicate selection will not impact either of these goals for improving the 510k process. In addition, companies do not select predicates only for the reasons indicated in this FDA predicate selection guidance. One of the most common reasons for selecting a predicate is the cost of purchasing samples of predicate devices for side-by-side performance testing. This only relates to cost, not safety or performance, and forcing companies to purchase more expensive devices for testing would not align with the least burdensome approach. Another flaw in this proposed additional information to be included in the 510k summary is that there is a huge variation in the number of predicates that can be selected for different product classifications. For example, 319 devices were cleared in the past 10 years for the FLL product classification (i.e., clinical electronic thermometer), while 35 devices were cleared in the past 10 years for the LCX product classification (i.e., pregnancy test). Therefore, the approach to selecting a predicate for these two product classifications would be significantly different due to the number of valid predicates to choose from. This makes it very difficult to create a predictable or consistent process for predicate selection across all product classifications. There may also be confidential, strategic reasons for predicate selection that would not be appropriate for a 510k summary.
Section 7 – Examples
The FDA predicate selection guidance provides three examples. In each example, the FDA is suggesting that the submitter should provide a table that lists the valid predicate devices and compare those devices in a table using the four best practices as criteria for the final selection. The FDA is positioning this as providing more transparency to the public, but this information presented in the way the FDA is presenting it would not be useful to the public. This is creating more documentation for companies to submit to the FDA without making devices safer or improving efficacy. This approach would be a change in the required content of a 510k summary and introduce post-market data as criteria for 510k clearance. This is a significant deviation from the current FDA policy.
Example 1 from predicate selection guidance
In this example, the submitter included a table in their 510k submission, along with their rationale for selecting one of the four potential predicates as the predicate device used to support their 510k submission. This example is the most concerning because the summary doesn’t have any details regarding the volume of sales for the potential predicates being evaluated. The number of adverse events and recalls is usually correlated with the volume of sales. The proposed table doesn’t account for this information.
Example 2 from predicate selection guidance
In this example, the submitter was only able to identify one potential, valid predicate device. The submitter provided a table showing that the predicate did not present concerns for three of the four best practices, but the predicate was the subject of a design-related recall. The submitter also explained the measures taken to reduce the risk of those safety concerns in the subject device. As stated above, using the occurrence of a recall as the basis for excluding a predicate is not necessarily appropriate. Most recalls are initiated due to reasons other than the design. Therefore, you need to make sure that the reason for the recall is design-related rather than a quality system compliance issue or a vendor quality issue.
Example 3 from predicate selection guidance
In this example, the submitter identified two potential, valid predicate devices. No safety concerns were identified using any of the four best practices, but the two potential devices have different market histories. One device has 15 years of history, and the second device has three years of history. The submitter chose the device with 15 years of history because the subject device had a longer regulatory history. The problem with this approach is that years since clearance is not an indication of regulatory history. A device can be cleared in 2008, but it might not be launched commercially until several years later. In addition, the number of devices used may be quite small for a small company. In contrast, if the product with three years since the 510k clearance is distributed by a major medical device company, there may be thousands of devices in use every year.
Medical Device Academy’s recommendations for predicate selection
The following information consists of recommendations our consulting firm provides to clients regarding predicate selection.
Try to use only one predicate (i.e., a primary predicate)
Once you have narrowed down a list of predicates, we generally recommend only using one of the options as a primary predicate and avoiding the use of a second predicate unless absolutely necessary. If you are unsure of whether a second predicate or reference device is needed, this is an excellent question to ask the FDA during a pre-submission teleconference under the topic of “regulatory strategy” (see image below). In your PreSTAR you can ask the following question, “[Your company name] is proposing to use [primary predicate] as a primary predicate. A) Does the FDA have any concerns with the predicate selection? B) Does the FDA feel that a secondary predicate or reference device is needed?”
When and how to use multiple predicates
Recently a client questioned me about the use of a secondary predicate in a 510k submission that I was preparing. They were under the impression that only one predicate was allowed for a 510k submission because the FDA considers the two predicate devices to be a “split predicate.” The video provided above explains the definition of a “split predicate,” and the definition refers to more than the use of two predicates. For many of the 510k submissions, we prepared and obtained clearance for used secondary predicates. An even more common strategy is to use a second device as a reference device. The second device may only have technological characteristics in common with the subject device, but the methods of safety and performance testing used can be adopted as objective performance standards for your 510k submission.
When you are trying to use multiple predicate devices to demonstrate substantial equivalence to your subject device in a 510k submission, you have three options for the correct use of multiple predicate devices:
Two predicates with different technological characteristics, but the same intended use.
A device with more than one intended use.
A device with more than one indication under the same intended use.
If you use “option 1”, then your subject device must have the technological characteristics of both predicate devices. For example, your device has Bluetooth capability, and it uses infrared technology to measure temperature, while one of the two predicates has Bluetooth but uses a thermistor, and the other predicate uses infrared measurement but does not have Bluetooth.
If you use “option 2”, you are combining the features of two different devices into one device. For example, one predicate device is used to measure temperature, and the other predicate device is used to measure blood pressure. Your device, however, can perform both functions. You might have chosen another multi-parameter monitor on the market as your predicate, however, you may not be able to do that if none of the multi-parameter monitors have the same combination of intended uses and technological characteristics. This scenario is quite common when a new technology is introduced for monitoring, and none of the multi-parameter monitors are using the new technology yet.
If you use “option 3”, you need to be careful that the ability of your subject device to be used for a second indication does not compromise the performance of the device for the first indication. For example, bone fixation plates are designed for the fixation of bone fractures. If the first indication is for long bones, and the second indication is for small bones in the wrist, the size and strength of the bone fixation plate may not be adequate for long bones, or the device may be too large for the wrist.

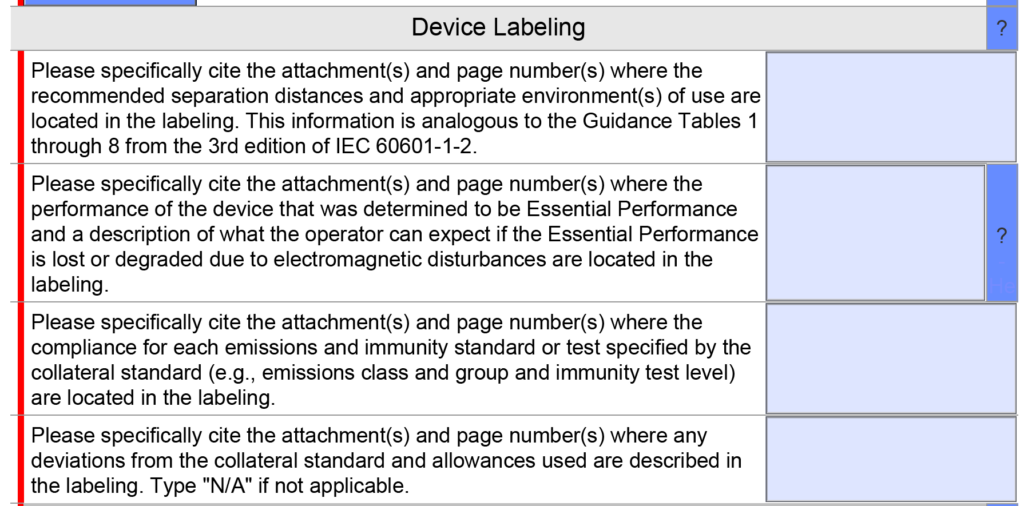

 template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device:
template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device: