MDUFA V is the agreement between the FDA and the medical device industry to fund the review of medical device submissions by the FDA.
What is MDUFA V?
The Medical Device User Fee and Modernization Act (MDUFMA or MDUFA) is a set of agreements between the Food and Drug Administration (FDA) and the medical device industry to provide funds for the Office of Device Evaluations (ODE) to review medical device submissions. FDA user fees were first authorized via MDUFMA in 2002 for FY 2003. Each MDUFA reauthorization has lasted five years, and FY 2023 is the 21st year.
How are the MDUFA V user fees decided?
Section 738A(b)(1) of the FD&C Act requires that the FDA consult with various stakeholders, including representatives from patient and consumer advocacy groups, healthcare professionals, and scientific and academic experts, to develop recommendations for the next MDUFA five-year cycle. The FDA initiated the reauthorization process by holding a public meeting on October 27, 2020, where stakeholders and other public members could present their views on the reauthorization. The following is a list of the four industry groups represented in the MDUFA V negotiations with the FDA:
The FD&C Act further requires that the FDA continue meeting with the representatives of patient and consumer advocacy groups at least once every month during negotiations with the regulated industry to continue discussing stakeholder views on the reauthorization and their suggestions for changes.
What are FDA user fees?
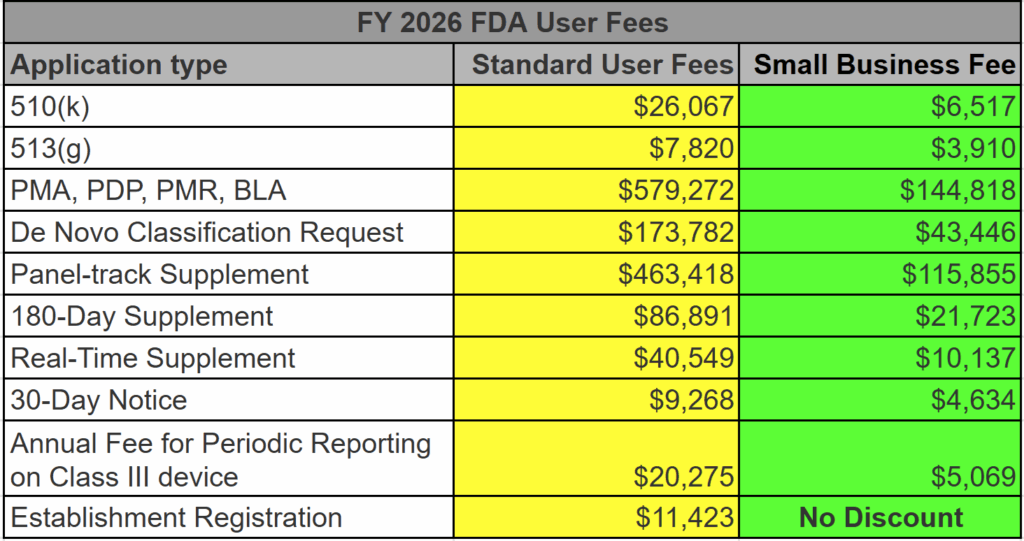
At the very core of it, the FDA user fees fund the FDA Office of Device Evaluation (ODE) budget. Without these user fees, the FDA cannot begin reviewing a medical device submission. This includes 510k, PMA, and De Novo submissions. Before the FDA assigns a reviewer to your submission, you must pay the appropriate device user fee in full unless eligible for a waiver or exemption. If you pay the user fee by credit card, you must allow a few extra days for the user fee to clear. Otherwise, your submission will be placed on “User Fee Hold.” Small businesses may qualify for a reduced fee. The FDA announced the FY 2026 FDA User Fees on July 31, 2025. The FDA will announce the user fees for FY 2027 in a Federal Register notice around August 2026.
When does MDUFA V take effect?
Our team regularly checked the announcement of the MDUFA V user fees from August until the October 5, 2022 announcement. The announcement of the FY 2023 user fees was delayed because Congress did not approve the MDUFA reauthorization until the last week of September. The new user fees were initially expected to take effect on October 1, 2022, but the announcement of actual user fees for 2022 was announced on October 5, 2022. This was two months later than expected.
Why was MDUFA V delayed, and will it happen again?
MDUFA V was delayed because the user fee reauthorization requires an act of Congress. The House of Representatives approved the Food and Drug Amendments of 2022 on June 8, 2022. However, the Senate did not file a bill until after the August recess. There were also differences between the legislation the House and the Senate proposed. Therefore, to ensure that the FDA did not have to furlough employees when MDUFA IV funding expired, the President approved and signed a temporary reauthorization on September 30, 2022. The short-term continuing resolution is a temporary stopgap to fund the FDA until December 16, 2022. However, the continuing resolution covers funding for medical device user fees through September 30, 2027. Therefore, the device industry can expect the FDA to continue to operate regardless of the outcome of temporary policies that expire this December. Still, similar delays occurred with previous MDUFA reauthorization, and we expect more of the same US partisan politics between August 2027 and the November 2027 election.
How much did MDUFA V user fees increase?
The increase is dependent upon the fee type. Annual registration fees are increasing by 14.47% (i.e., $5,672 to $6,493). The MDUFA V user fees increased by a stupendous amount (+55.90%) from $12,745 to $19,870 for the 510k user fees. Yikes! De Novo Classification Requests increased by 17.79% from $112,457 to $132,464. Other submissions increased by similar amounts. For more details, check out the table below (also posted on our homepage).
FDA User Fee FY 2023 represents a 55.90% increase in the 510(k) user fee
Do user fees ever decrease?
If we lived in a magical world where gas prices dropped and stayed low, the inflation-adjusted pricing would decrease for FDA user fees. That has happened once, but I fit into skinny jeans once too. The increase in FDA user fees from FY 2023 to FY 2024 was 9.5%, except the Annual Registration Fee, which increased by 17.87% to $7,653.
Why is August 1st important?
August 1st is the first day the FDA acceptsSmall Business Certification Requests for the new fiscal year. That means any small business that wants to keep small business status needs to reapply, and any new business that qualifies for small business status must also apply. The importance of applying for small business status is how much you could save on your submission. The FDA will complete its review of the Small Business Certification Request within 60 calendar days of receipt. Upon completion of the review by the FDA, the FDA will send you a decision letter with your small business designation number or a justification for denial.
Does small business status expire?
Yes, small business status expires. The small business status expires on September 30 of the fiscal year it is granted. A new MDUFA Small Business Certification Request must be submitted and approved each fiscal year to qualify as a small business. If you forget to reapply for small business status on August 1, you can reapply anytime during the year. Still, you will temporarily lose small business status from October 1 until the qualification is renewed. The good news is there is no fee associated with submitting a Small Business Certification Request. For more information, please visit our webpage dedicated to small business qualifications.
The format and content requirements for an FDA pre-submission have not changed, but the launch of the FDA PreSTAR has changed everything.
What is an FDA pre-submission?
An FDA pre-submission aims to get answers to questions you have about a future FDA submission. The pre-submission may consist of one large PDF document or multiple PDF documents. In your pre-submission, you must select either an email response or an email response with a teleconference. One advantage of choosing a teleconference is that you can ask clarifying questions during a one-hour teleconference with the FDA. Still, you are responsible for submitting draft meeting minutes to the FDA within 15 days of the teleconference. If you select an email response, you do not need to provide meeting minutes to the FDA.
Our new 4-part FDA pre-submission webinar series is available on-demand. You can download it and watch it as many times as you want. This will be the Ultimate FDA pre-submission training. Do not miss it.
Everyone asks us for examples, so we will show you how to complete the entire FDA PreSTAR for a device in this webinar series. If you would like to vote on which device we should use as an example (i.e., Option 1 = Infrared Thermometer or Option 2 = Antimicrobial Gauze), please place your vote on our LinkedIn page.
What is the difference between an FDA pre-submission and a Q-submission?
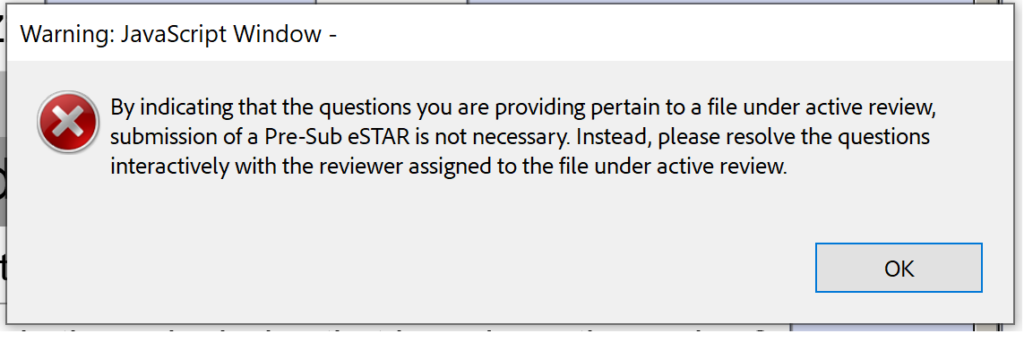
Every FDA pre-submission is a Q-submission, but not all Q-submissions are pre-submissions. The new PreSTAR template is currently limited to an FDA pre-submission, but the template will be expanded to other types of Q-subs later. The FDA pre-submission template (i.e., PreSTAR) beta version 0.1 is unnecessary for responses to interactive review questions from the FDA. Just email the Lead Reviewer (file size limit is 25 MB for email).
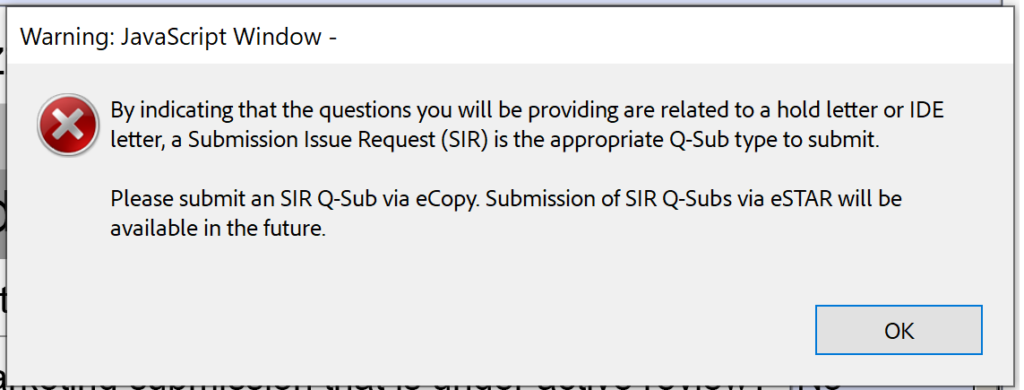
Unfortunately, the beta version 0.1 is also not ready for Submission-in-Review (SIR) meetings or responses to IDE during an interactive review.
13 other types of submissions might benefit from Q-submissions:
Submission Issue Requests (SIRs)
Study Risk Determinations
Informational Meetings
Breakthrough Device Designation Requests
Informational Meetings
PMA Day 100 meetings
Agreement and Determination meetings
Submissions associated with the STeP program
Accessory classification requests
Requests for FDA feedback on specific questions or cross-cutting policy matters
Requests for recognition of publicly accessible genetic variant databases
Combination product agreement meetings (CPAM), and
Feedback on FDA 483 inspection observations.
We expect the PreSTAR template to eventually be available for a 513(g) request in the future because it was already validated for that purpose.
What is the Q-submission number?
All Q-submissions are assigned a document number beginning with “Q” upon receipt (i.e., Qyyxxxx). The format of the number consists of 2-digits (i.e., “yy”) for the year of submission (e.g., “23” for 2023) and 4-digits (i.e., “xxxx”) that are the following sequential number assigned by the FDA for that calendar year. Therefore, the first Q-submission received by the FDA in January 2023 is Q230001, and between 3,500 and 4,000 new submissions are usually received each year. If the subject device was submitted in a previous Q-submission, the original document number is re-used, and a supplement number is added (i.e., Qyyxxx/S001, Qyyxxx/S002, etc.). Q-submission numbering is explained in more detail in the 2023 FDA guidance.
Does the FDA charge for Q-submissions?
FDA pre-submissions do not require paying an FDA User Fee (i.e., $0).
How long does an FDA pre-submission take?
The days of squishing timelines are gone. The timeline is 70-75 calendar days. On October 5, 2022, MDUFA V was approved. As one of the MDUFA V decision targets, the FDA is tasked with reducing the timeline for responding to pre-sub questions within 70 days for 90% of pre-sub requests. The FDA is tasked with achieving this goal by March 2024. If they are successful, the FDA will receive an increase of 59 headcounts to their budget in 2024. This is approximately a $19 million incentive to respond to your pre-submission meeting questions within 70 days. To reflect these new MDUFA V decision targets, the FDA updated the Q-Sub guidance document to reflect the target date of 70 days for the email response and 75 days for teleconference meetings. The FDA also updated the Customer Collaboration Portal (CCP) to facilitate tracking of FDA pre-submission deadlines.
What is an FDA PreSTAR?
In the past, you had to create your document(s) for an FDA pre-submission. Some people create one large PDF document divided into sections, while others create separate PDF documents for each requirement of the FDA pre-submission guidance. On August 14, 2023, the FDA released an updated beta version (i.e., version 0.2) of a new PDF template (i.e., FDA PreSTAR). This new PreSTAR template provides multiple benefits to the FDA:
every company uses the same format,
the template automatically verifies that the pre-submission includes all required elements, and
Including optional elements will encourage companies to provide more device details than they might otherwise provide.
The PreSTAR also benefits submitters:
you will never forget the required elements of the FDA pre-submission,
you never have to validate an FDA eCopy, and
the similar format and user interface will train you to use the FDA eSTAR.
Note: October 1, 2023, was the FDA eSTAR implementation deadline.
Do you have to use the PreSTAR template?
Nope. The PreSTAR version 0.2 is a beta version and 100% optional. However, I like it better than my templates. Your design team can still have individual documents for the user manual, device description, and testing plan. We attach the document using the button that says “Add Attachment” (see screen capture below).
The PreSTAR template was built by Patrick Macatangga, a Tools & Templates Engineer working at the FDA on the Lifecycle Tools and Templates Team. To help with where to direct questions about the template, he suggested:
If you have questions or feedback regarding the voluntary use of the eSTAR for medical devices regulated by CDRH, or if you have general questions about medical devices, contact the Division of Industry and Consumer Education (DICE).
If you find malfunctions or errors in the eSTAR template for medical devices regulated by CDRH, contact eSubPilot@fda.hhs.gov.
If you have questions regarding 510(k)s, De Novo requests, or Early Submission Requests for medical devices regulated by CDRH, contact OPEQSubmissionSupport@fda.hhs.gov.
How do you submit an eCopy?
You can submit an FDA eCopy on electronic media (e.g., USB flash drive) and send it via FedEx to the FDA Document Center at the following address: Food and Drug Administration, Center for Devices and Radiological Health, Document Mail Center, 10903 New Hampshire Ave., Bldg. 66, rm. G609, Silver Spring, MD 20993-0002. However, you can also submit an FDA eCopy via a web browser (i.e., CCP…see next section on submitting a PreSTAR).
If you are submitting an eCopy through the CCP instead of an FDA PreSTAR
How do you submit a PreSTAR?
You have two options for delivery of an FDA pre-submission:
save the pre-sub on electronic media (e.g., USB flash drive) and send it via FedEx to the FDA, and
upload the pre-sub to the new FDA Customer Collaboration Portal (CCP).
As you can guess from the video above, we only use option 2 for FDA pre-submissions. For option 2, you can upload an eCopy (saved as a zip file) or a PreSTAR (in the native PDF format). The image below shows you how this is done, but the uploading process usually takes about one minute–depending on your file size and bandwidth. You can register for your own CCP account in seconds.
What is the pre-submission process?
Preparing and uploading your FDA pre-submission meeting request is only the first step of the process. You will receive an automated email confirming that your pre-submission was successfully uploaded. Then, you will receive an automated letter via email that gives you the assigned Q-sub number. You will also receive an automated email notifying you that the pre-submission was accepted, or the FDA reviewer will contact you if changes are needed. The FDA reviewer assigned will usually contact you by email within the first three weeks to schedule a teleconference if you request one. Still, the date/time offered usually does not match the availability of the FDA team, and alternate dates/times may be offered.
You will receive an email response from the FDA for each of your questions within 70 days of receipt by the FDA. If you requested a teleconference, it would typically be about 75 after receipt of the FDA pre-submission meeting request. Your team needs to prepare a detailed discussion plan for the one-hour teleconference. A slide deck is highly recommended to facilitate communication but is not required. If you provide a slide deck, you should email it to the reviewer before the meeting. You must also provide a copy of the slide deck with your meeting minutes. At the beginning of the teleconference, someone from your team must commit to submitting draft meeting minutes to the FDA within 15 days. The FDA will reply with acceptance of your meeting minutes, or they will provide an edited version. It is also common to submit a supplement FDA pre-submission with detailed protocols and new questions for the FDA.
Everyone has a favorite resource they use to answer regulatory questions, but can you trust OpenAI or Elsmar to answer correctly?
If you are deathly afraid of trying new technology, the image above is a screen capture from OpenAI describing “itself.” OpenAI is artificial intelligence (AI), but it is not self-aware yet. The image below is a screen capture from the “About” webpage for Elsmar Cove. This article was the oldest post on the Medical Device Academy, and it described how to use Elsmar Cove as a resource for quality systems and regulatory questions. To update that blog, we are comparing the use of OpenAI with Elsmar Cove. Just in case you were wondering, Elsmar Cove is #6 on our list of favorite search tools, and OpenAI is #5:
Search the FDA.gov device databases – 3rd place, you can email DICE or call 301.796.7100
Search Google (world’s #1 search engine) – 4th place, ask any 5-year-old for help
Are the answers provided by OpenAI and Elsmar Cove accurate?
To test the accuracy of a common regulatory question, we chose a question we weren’t 100% sure about when a client asked last month. I asked my team, but nobody was 100% certain. Basil Systems is limited to submission and post-market surveillance data. I searched FDA.gov, but it was not clear. Google gave us a link to the FDA website. I asked a couple of ex-FDA consultants, but they gave me outdated information. On Thursday, March 30, 2023, I asked Lisa King during an AAMI course I was co-teaching. Lisa is a Consumer Safety Officer at the FDA responsible for reviewing device entries into the FDA. She is also in very high demand for public training courses. She said the contract manufacturers used to be exempt from registration if they shipped to a legal manufacturer first. The regulations changed, and now 100% of contract manufacturers making a finished device must register with the FDA. She also clarified that the FDA doesn’t use the term “legal manufacturer.”
As you can see from the above answer provided by OpenAI, the ChatGPT engine [i.e., Model: Default (GPT-3.5)] effectively produces the correct answer. Using the same wording for the regulatory question, “Does a foreign contract manufacturer need to register with the FDA if they are shipping the medical device to the legal manufacturer first before the device is exported to the USA?” there were no results from Elsmar Cove. After several attempts, I found what I was looking for using the following search terms, “FDA registration of contract manufacturers.” There were multiple related search results, but the most useful discussion threads in the Elsmar Cove discussion forum were:
The most succinct correct answer in the forum is copied below.
Can you trust OpenAI and Elsmar Cove to answer your regulatory questions?
OpenAI is only as effective as the data used to train it. This is constantly evolving, but we have identified search results that were 100% accurate, results that were outdated, and results that were scary wrong. The same is true of discussion forums. Elsmar Cove is one of the best discussion forums for the medical device industry, but people also use ASQ, RAPS, and AAMI. The quality of the information provided depends upon the knowledge and experience of the people participating in the forum, but it also depends upon the forum’s moderation. Elsmar Cove has some experienced moderators with decades of experience. There is always the chance that the most experienced person in the world could answer your regulatory question incorrectly. This usually creates a problem because everyone else in the forum hesitates to challenge a recognized expert. Therefore, regardless of which resource(s) you use, always try to get a reference to the trustworthy source of the applicable regulation. Even Lisa King could make a mistake, but she immediately said, you can find the regulations in the US Code of Federal Regulations (i.e., 21 CFR 807). The bottom line is, always do your fact-checking and reference your source(s).
It’s a common misconception that FDA De Novo content is very different from FDA 510k submission content, but is that true?
What do you think the De Novo content differences are?
Most people think the difference between a 510k and a De Novo is time and money. That conclusion is based upon a very important assumption: a 510k will not require clinical data, and a De Novo will require clinical data. That assumption is not always correct. 10-15% of 510k submissions include clinical data to support the performance claims, and last year our team submitted three De Novo submissions that did not include any clinical data. So what are the differences between a 510k and a De Novo content?
We use the same FDA eSTAR template for both types of FDA submissions, and on the first page of the eSTAR template, we identify if the submission is a 510k or De Novo. If we select De Novo, the eSTAR will be pre-populated with six unique De Novo content requirements covering four (4) different areas that are not found in a 510k. The six unique content requirements are:
Recommending a classification, providing a justification for that classification, and explaining what efforts were taken to identify a suitable 510k product code
Description of existing alternative practices or procedures used in diagnosing, treating, preventing, curing, or mitigating the disease or condition for which the IVD or device is intended
Providing a written benefit/risk analysis starting with the clinical benefits of your device
Efforts to identify a potential predicate (including identifying alternative practices, procedures, or even drugs)
Recommendations for FDA special controls for your new product code based upon the risks to health and the mitigation measures for each risk
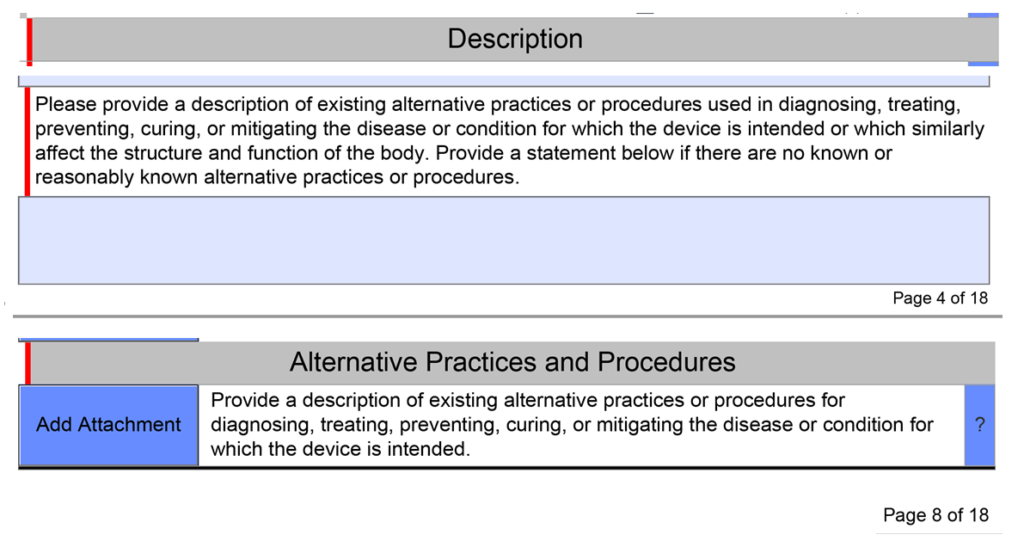
What alternative practices and procedures are currently available?
The unique De Novo content requirement is to provide a description of alternative practices and procedures for treatment or diagnosis of the same indications that you are proposing for your subject device. This is a subsection of the device description section in the FDA eSTAR template. Your should description should include other 510k-cleared products, drugs, and even products that have similar indications but are not identical. The description of alternative practices and procedures must also be attached as a document in the section for benefits, risks, and mitigation measures. To maintain consistency throughout your submission, you should create the document for attachment first and copy and paste the content into the text box at the end of the device description section.
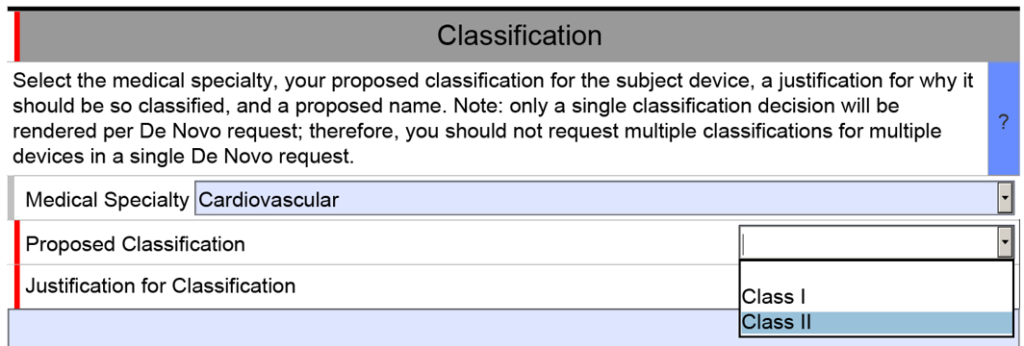
You need to recommend a classification in your De Novo
The unique De Novo content requirement is found in a section titled “Classification.” There is a shorter classification section included in 510k submissions, but the 510k version only has four cells. The first three are populated by selecting one of the options from a dropdown menu, and the fourth cell is only used if your subject device includes other product classification codes.
The De Novo version of the eSTAR is identical for the first row of the classification section, but then you must select a proposed product classification (i.e., Class 1 or Class 2) in accordance with FDA Classification Procedures (i.e., 21 CFR 860). The third cell is a text box for you to enter your justification for the proposed classification. Next, the FDA requires you to enter a proposed classification name. Finally, at the end of the classification section, the FDA requires that you provide a classification summary or reference to a previous NSE 510k submission.
A benefit-risk analysis is required in the De Novo content
For new devices, the FDA uses a benefit-risk analysis to decide if a device should be authorized for marketing in the USA. This process includes humanitarian device exemptions, De Novo applications, and Premarket Approval submissions. The FDA has a guidance document that provides guidance for FDA reviewers and the industry. The most important aspect is, to begin with, the benefits of the device and to provide a quantitative comparison of benefits and risks. Many De Novo submissions have been rejected because the submitter did not provide objective evidence of clinical benefits for the subject device.
The FDA guidance documents are helpful for creating a benefit-risk analysis, but you can also find information in the ISO/TIR 24971:2020–the guidance for the application of ISO 14971:2019. Our company also includes a template for a benefit/risk analysis as part of our risk management procedure (i.e., SYS-010).
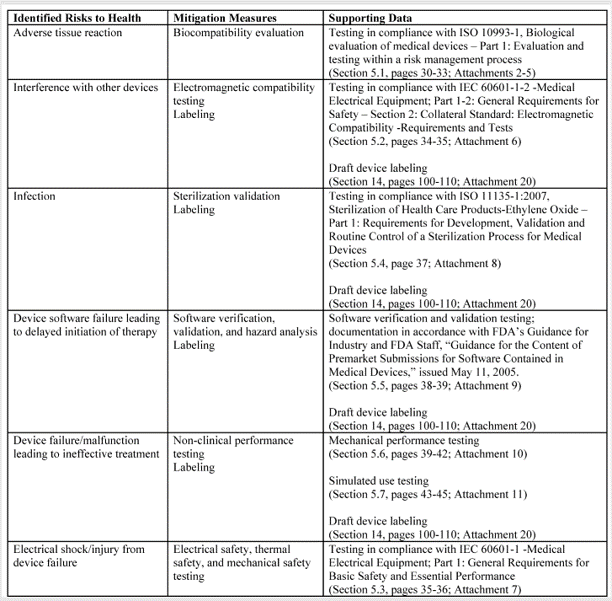
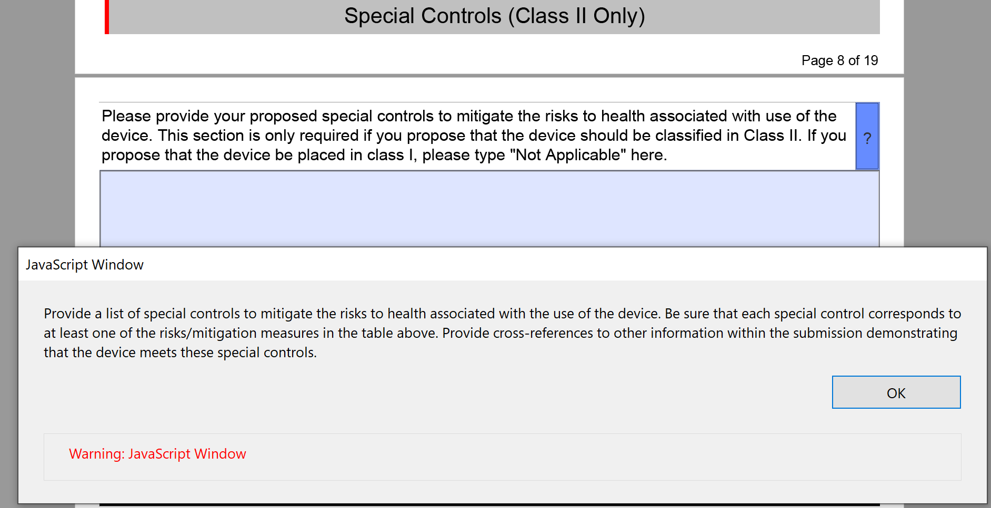
What are your recommended Special Controls?
In FDA De Novo Classification Decision Summaries, there is a table provided that identifies the identified risks to health and the recommended mitigation measures for each risk category. In the FDA eSTAR, you are required to add a similar table for De Novo content. The only difference between the table in summary and the eSTAR is that the eSTAR table has a third column where the FDA wants you to reference the supporting data provided for each mitigation measure–including the document and page within the document. The FDA also provided an example table in the eSTAR, copied below.
The above table for the risks to health and mitigations needs to be translated into a list of recommended Special Controls for Class II devices. Since most De Novo applications are for Class II devices, you will need to convert each of your mitigations into a corresponding Special Control and type these controls into the text box provided in the FDA eSTAR.
What else is different from a 510k?
There are no additional mandatory elements that you need to include in a De Novo application, but there are several elements of a 510k submission that are not included in a De Novo. The most obvious of these sections is the Substantial Equivalence Comparison Table in the section labeled “Predicates and Substantial Equivalence.” Another difference is that you are more likely to need clinical data to support a De Novo application than for a 510k submission. It is also possible that subsequent 510k submissions for the same product code may not need to provide clinical data because the 510k process only requires a demonstration of substantial equivalence rather than clinical benefits outweighing risks to health. The FDA review time for a Traditional 510(k) varied between 190 and 210 days in 2022, while the De Novo review timeline averaged 390 days in 2022. Finally, the FDA user fees for 510k submissions are far less than those for a De Novo application.
The FDA CCP keeps getting better. Now, you can request small business determination (SBD) online through the FDA CDRH portal.
Preparing your FDA eSubmission for the FDA CCP
There are three possible formats for an FDA eSubmission to the FDA CCP for CDRH: 1) FDA eCopy, 2) FDA eSTAR, and 3) Small Business Determination (SBD) Request. We explain each in sequence below:
FDA eCopy:
The FDA eCopy is no longer an acceptable format for a 510k submission. You must use the FDA eSTAR template for a 510k submission. In contrast, pre-submission meeting minutes and withdrawal letters require an FDA eCopy. An FDA eCopy is also required for Sprints (i.e., pre-sub variation for Breakthrough devices and STeP devices). For a De Novo Submission and 513(g) submissions, you have the option of using the FDA eSTAR or the FDA eCopy (BUT use the FDA eSTAR or PreSTAR respectively). Here are the preparation steps:
Confirm your eSTAR is complete. There are three ways to know that your eSTAR is complete and ready for upload, but don’t worry too much. The FDA validated that the FDA CCP will automatically detect an incomplete eSTAR and will not allow it to be uploaded.
Bold Font at the top of the eSTAR or PreSTAR template is Green
All of the color-coded bars on the left side of the template are Green or Grey
All of the sections of the template in the verification section are found on the left side
Small Business Determination Request:
The ability to upload your small business qualification documents as an eSubmission instead of sending a hardcopy via courier is a new feature enabled by the FDA on Saturday night (i.e., September 29, 2024). Confirm your eCopy complies with FDA’s eCopy guidance.
#1 Sign in to the portal on the login page. If you don’t already have your own account, you can Sign up in less than 5 minutes.
#2 Accept the FDA User Terms and Conditions
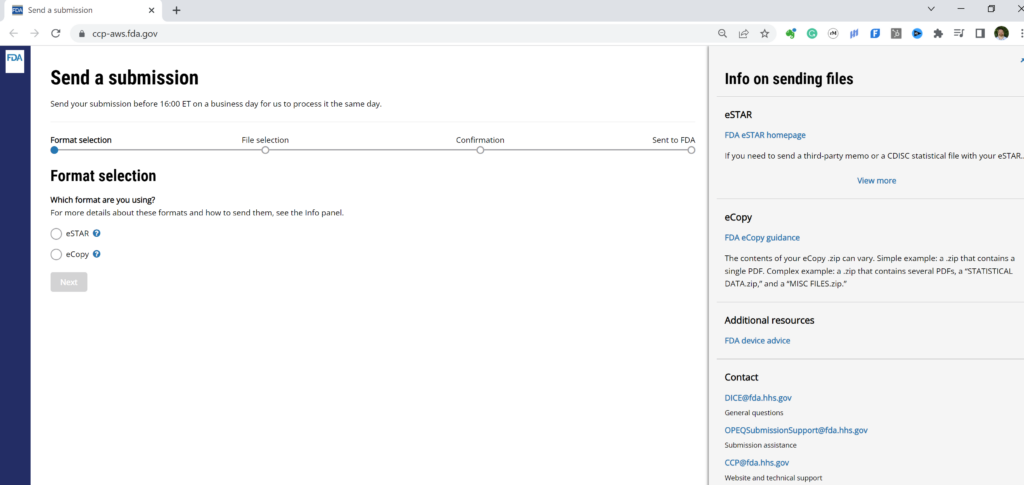
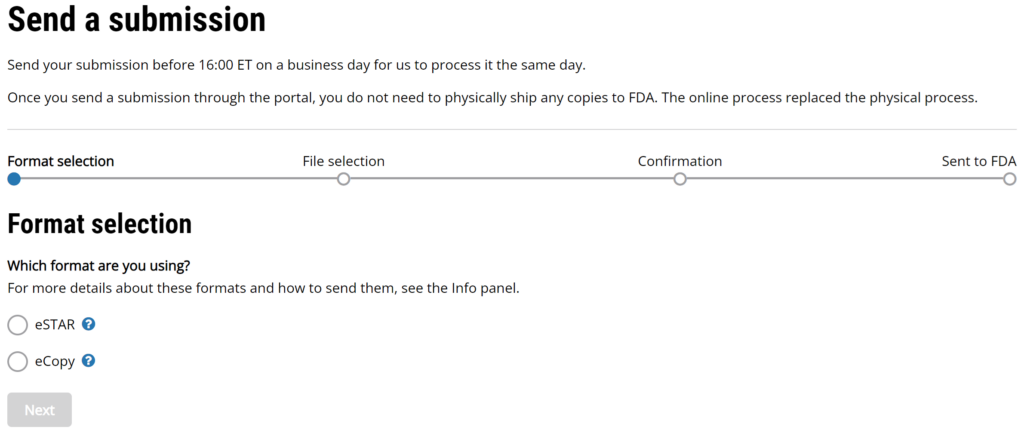
#3 You have three options one you accept the FDA user terms and conditions. One option is to check on the status of prior eSubmissions on the Home page. Your second option is to send a new FDA eCopy or FDA eSTAR. For this second option you click on the “+” symbol on the left panel of the webpage (if you hover over the “+” symbol, you will see “Send a submission”) or you can expand the left panel as I have done in the picture below. The third option (i.e., the newest FY2025 option) is to create a request for small business determination (i.e., SBD).
#4 If you selected “Send a submission” you can upload your FDA eCopy or FDA eSTAR. The top of the page has the following instructions “Send your submission before 16:00 ET on a business day for us to process it the same day. Once you send a submission through the portal, you do not need to physically ship any copies to FDA. The online process replaced the physical process.”
#5 Click on the “Next” button that appears below the selection formats once a format is selected.
#6 Drag & drop your single “.zip” file here, or browse for it.
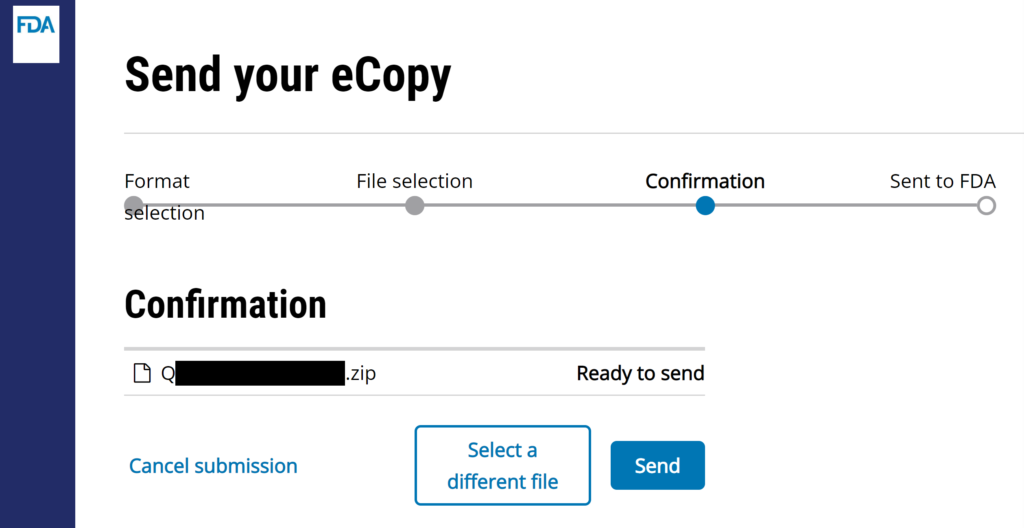
#7 The webpage will ask you for a short description of your submission. The company name and type of submission is sufficient [e.g., “Medical Device Academy – presubmission meeting minutes (i.e., Q24xxxx.A001)”]. Click on “Send” button to complete the uploading process.
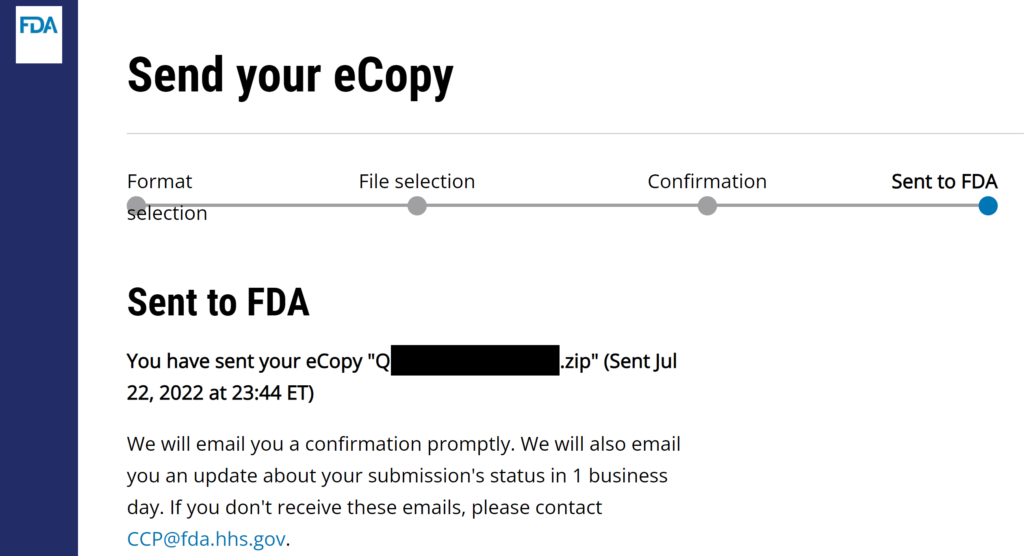
#8 Verify that the FDA CCP site gives you a confirmation for the successful uploading of your submission. I always create a screen capture of the confirmation page once I hit send, but that is unnecessary but faster to insert in an email to your boss. You will receive an email seconds later.
The process is identical for the FDA eSTAR, but you DO NOT need to zip an FDA eSTAR or PreSTAR.
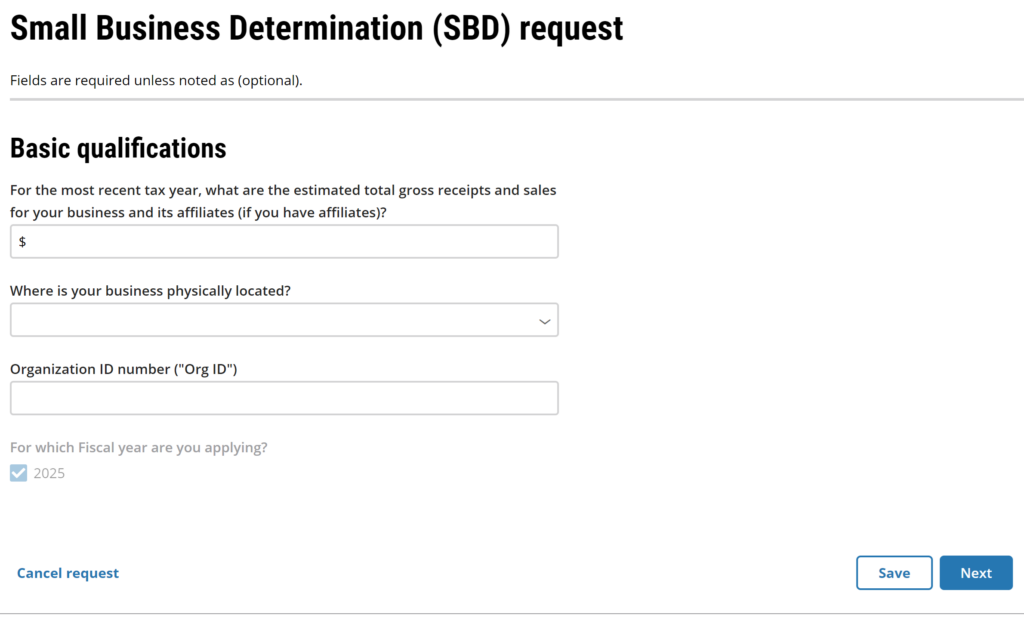
Starting a Small Business Determination (SBD) Request
The third type of submission for the FDA CCP is a small business determination. This new option was enabled last Saturday night (i.e., September 28, 2024)….what a thoughtful birthday present from the FDA to me. Just click on the document icon beneath the plus sign. The website will take you to the “Create a request” page where you can begin your SBD request. Before you start this process, you will need to prepare some documents: #1 Tax return for the most recent tax year #2 Organization ID number (watch our YouTube Video)
July 2022 Update for the FDA eCopy process (FDA CCP is launched!)
Finally, we can use the new FDA CCP to eliminate FedEx shipments, and 100% of your submissions will be electronic through the portal. The FDA created a Customer Collaboration Portal (CCP) for medical device manufacturers. Initially, the portal’s purpose was to provide a place where submitters could track the status of their submissions and verify the deadlines for each stage of the submission review process. On July 19, 2022, the FDA emailed all active FDA CCP account holders that they can upload both FDA eCopy and FDA eSTAR files to the portal 100% electronically. The FDA released a draft eSTAR guidance as well. Since our consulting team sends out submissions daily, everyone on the team was able to test the new process. If you have a CCP account, you no longer need to ship submissions via FedEx to the Document Control Center (DCC).
FDA Q&A about the new FDA CCP Submission Uploading Process
Medical Device Academy Question: Who will be permitted to use the FDA CCP to upload submissions for the DCC? FDA Response: We will first offer this feature in batches to people like you who already use CCP so we can study its performance. We will then refine it and make it available to all premarket submitters.
Medical Device Academy Question: What do you need to use the FDA CCP? FDA Response: You don’t need to do anything to participate since you already use CCP. We will email you again when you can start sending your next submissions online.
Medical Device Academy Question: Suppose another consultant asks me to submit an eSTAR or eCopy for them, or I do this for a member of my consulting team. Is there any reason I cannot upload the submission using my account even though the other person is the official submission correspondent and their name is listed on the cover letter? FDA Response: The applicant and correspondent information of the submission is still used when logging the submission in. The submitter (i.e., the person uploading the submission) is not used in any part of the log-in process. The submission portal is essentially replacing snail mail only; once the DCC loads the submission, whether it be from a CD or an online source, the subsequent process is identical to what it used to be, for now.
Medical Device Academy Question: Is there any type of eCopy that would not be appropriate for this electronic submission process (e.g., withdrawal letters, MAF, or breakthrough device designations)? FDA Response: You can use the eCopy option to submit anything that goes to the DCC, so all your examples are fair game, though interactive review responses would still be emailed to the reviewer.
Medical Device Academy Question: How can I get help from the FDA? FDA Response: If you have questions, contact us at CCP@fda.hhs.gov.
Before you complete FDA forms for your 510k submission, you need to made sure you have the most updated FDA forms.
How do you know if the FDA form you are using is current?
The FDA assigns numbers to each FDA form and the document control number is found in the bottom left footer of the document. In addition, the top right-hand header of the document will have an expiration date for the form (see the picture below). Often the changes to FDA forms are minor, but you should only submit the current version of the FDA form which has not expired.
What happens if you are using an expired FDA form?
In the past, if you included an obsolete document in your submission the FDA would often ignore this an proceed with the review of your submission anyway. Now FDA reviewers will identify the obsolete form and require you to resubmit the document on the current version of the form. If the reviewer is conducting an initial Refusal to Accept (RTA) screening, and one of the required items in the RTA screening are identified, then you will receive an RTA Hold letter and the RTA checklist will include a comment that you have used an obsolete version of an FDA Form.
If there are no deficiencies identified in the RTA checklist, the reviewer may still send you an email asking you to submit the document on the correct form. This could be a formal amendment (e.g. K123456/A001) or it could be as an informal email of the corrected document. This type of request could also be identified after the substantive review is complete in the form of a comment in an Additional Information (AI) Request or as part of an Interactive Review Request. An AI Request must be responded to with a formal supplement submitted to the Document Control Center (DCC) as a supplement to the original submission (e.g. K123456/S001) or as an informal ammendment submitted by email.
Examples of updated FDA forms for your 510k submission
Expired forms are frequently submitted to the FDA because submitters are using templates that have not been properly maintained or the submitter modified a form that was submitted in a previous 510k submission. The most common examples include: FDA Form 3514 (i.e. Submission Coversheet), FDA Form 3881 (i.e. Indications for Use), and the RTA Checklist.
Where can you find updated FDA forms?
Recently one of our clients noticed that the 510k template folder we share with people that have purchased our 510k courseincluded obsolete templates for Financial Disclosure. There are three financial disclosure forms that can be used for a 510k submission or De Novo Classification Request:
FDA Form 3454, Certification: Financial Interest and Arrangements of Clinical Investigator (PDF)
FDA Form 3455, Disclosure: Financial Interest and Arrangements of Clinical Investigators (PDF)
FDA Form 3674, Certification of Compliance, under 42 U.S.C. , 282(j)(5)(B), with Requirements of ClinicalTrials.gov (PDF)
We normally update these FDA forms as soon as the new form is released, but this financial disclosure forms are only used in about 10-15% of 510k submissions.
The current version of most FDA forms can usually be found by simply conducting an internet search for the form using your favorite browser. However, sometimes you may find a copy of the document that was editted by a consultant to facilitate completion of the document as an unsecured PDF or Word document. Although this is convenient, you should not use these “bastardized” forms. You should use the original secured form provided by the FDA. These native forms require Adobe Acrobat to complete the form and save the content. The most current version of the FDA form can be found using the FDA’s Form search tool.
Editing and Signing FDA Forms
Most of the FDA forms are secured and you can only enter information in specific locations. If there is a location for a signature, usually the signature cannot be added in Adobe to the secured form. In these situations, our team will save the document as a “Microsoft Print PDF” format. Once the document has been saved in this “non-native” format, you can manipulate almost anything in the document. Then we will add signatures using the “Fill and Sign” tool in Adobe Acrobat or we will use the “Edit” tool. Editing also gives us ability to make corrections when the document has incorrect information filled in the form somewhere.
Another option for adding dates and signatures is for you to save the document as a non-secure PDF. Then using an electronic signature software tool like Docusign, you can request that another person add their electronic signature or you can add your own electronic signature. Some companies prefer to do this to ensure the electronic signature meets 21 CFR Part 11 requirements, but the FDA accepts scanned images of a signature that was added to the document without certification in a 510k submission. This is even true for the Truthful and Accuracy Statement for a 510k. That document can be attached as a PDF in an FDA eSTAR template or you can electronically sign the eSTAR template if the person preparing the eSTAR is also the person signing the Truthful and Accuracy Statement.
Tips and Tricks for maintaining templates
Our company is a consulting firm, and we do not have a formal document control process that would be typical of our clients. However, we do have a shared Dropbox folder where we maintain the most current version of 510k templates. Any obsolete versions we move to an archive folder. However, there are ways to improve this informal system. You can include a date of the document in the file name. For example, “Vol 4 001_Indications for Use (FDA Form 3881) rvp 2-7-2022.” This indicates that this file is the FDA Form 3881 which is the indications for use form used in Volume 4 of the 510k submission. The document is the first document in that volume. The date the form was revised and saved is February 7, 2022 and the author’s initials are “rvp.”
If you are saving 510k templates you might consider adding an expiration date to the file name. For example, “Vol 4 001_Indications for Use (FDA Form 3881) exp 06-30-2023.” This file name indicates that the form’s expiration date is June 30, 2023. The inclusion of an expiration date in the file name is a visual reminder of when you will need to search for an updated FDA form.
A third way to manage your FDA Forms is to include them in your documents of external origin. ISO 13485:2016, Clause 4.2.4, requires that you maintain control of documents of external origin. Therefore, if your company has a formal quality system, a list or log of documents of external origin is the best way to manage FDA forms. Your log should indicate the date the updated FDA form was created, any parent guidance documents should be cross-referenced, and the expiration date of the FDA form should be identified. By using a log of this type, you can sort the list by expiration date or by the date of creation if there is no expiration date identified. Sorting the list will help your team prioritize which documents need to be reviewed next for new and revised versions.
Additional 510k submission resources
The FDA will be updating the 510k guidance for the new FDA eSTAR template by September 2022. Medical Device Academy will be systematically updating all of our templates and training webinars related to preparation of 510k submissions. We will also be preparing for the transition from FDA eCopy submissions to electronic submissions via a Webtrader Account.
You can keep up-to-date on template revisions in one of two ways:
Purchase our 510k course, and you will receive access to the updated templates as they are created. We will send email notifications each time a template is updated.
Register for our New Blog email subscription for automated email notifications of when a new blog is released about updated FDA forms, templates, and webinars.
Register for our New Webinar email subscription for automated email notifications of when a new or revised webinar is scheduled and for email notification of our newest live streaming YouTube videos.
I hated the the FDA eSubmitter template which was discontinued May 30, 2021. Finally we have eSTAR draft guidance for the new eSTAR template. Note: the final FDA eSTAR guidance was released on October 2, 2023 and we published a new blog the day of release.
History of 510k electronic submissions
The FDA has experimented with a multitude of pilot 510k submission programs over the years to streamline and improve the 510k submission content, formatting, and to facilitate a faster review process. The Turbo 510k program was one of the first successful pilot programs. In 2012, I wrote one of my first blogs about how to improve the 510k process. In September 2018, the FDA launched the “Quality in 510k Review Program Pilot” for certain devices using the eSubmitter electronic submission template. The goal of the this pilot program was to enable electronic submissions instead of requiring manufacturers to deliver USB flash drives to the FDA Document Control Center (DCC). I hated the eSubmitter template, and the FDA finally discontinued availability of the eSubmitter template on May 30, 2021. During the past 15 years, the FDA gradually streamlined the eCopy process too. Originally we had to submit one complete hardcopy, averaging 1,200 pages per submission, and one CD containing an electronic “eCopy.” Today, the current process involves a single USB flash drive and a 2-page printed cover letter, but today’s eCopy must still be shipped by mail or courier to the DCC.
eSTAR Pilot Program is Launched
During the 15-year evolution of the FDA eCopy, CDRH was trying to develop a reliable process for electronic submissions of a 510k. CBER, the biologics division of the FDA, has already eliminated the submission of eCopy submissions and now 100% of biologics submissions must be submitted through an electronic submissions gateway (ESG). In February 2020, CDRH launched a new and improved 510k template through the electronic Submission Template And Resource (eSTAR) Pilot Program. The eSTAR templates include benefits of the deceased eSubmitter template, but CDRH has incorporated additional benefits:
the templates use Adobe Acrobat Pro instead of a proprietary application requiring training;
support for images and messages with hyperlinks;
support for creation of Supplements and Amendments;
availability for use on mobile devices as a dynamic PDF;
ability to add comments to the PDF; and
the content and logic mirrors checklists used by CDRH reviewers.
Medical Device Academy’s experience with the eSTAR Templates
Every time the FDA has released a new template for electronic submissions we have obtained a copy and tried populating the template with content from one of our 510k submissions. Unfortunately, all of the templates have been slower to populate that the Word document templates that our company uses every day. On May 16 we conducted an internal training for our team on the eSTAR submission templates, and we published that training as a YouTube Video (see embedded video below). Then nine days later the FDA released updates to the eSTAR templates (version 0.7). The new eSTAR templates are available for non-IVD and IVD products (ver 0.7 updated May 27, 2021).
Sharon Morrow submitted our first eSTAR template to the FDA in August and we experienced no delays with the 510k submission during the initial uploading to the CDHR database, there was no RTA screening process, and CDRH did not identify any issues during their technical screening process. Shoron’s first eSTAR submission is now in interactive review, which is a better outcome than 95%+ of our 510k submissions. I have several other eSTAR submissions that are almost ready to submit as well. The other 510k consultants on our team are also working on their first eSTAR submissions.
Finally the CDRH releases an FDA eSTAR draft guidance
On September 29, 2021 the FDA released the new eSTAR draft Guidance for 510k submissions. This is a huge milestone because there have not been any draft guidance documents created for pilot programs. The draft indicates that the comment period will last 60 days (i.e. until November 28, 2021). However, the draft also states that the guidance will not be finalized until a date for requiring electronic submissions (i.e. submission via an ESG) is identified. The draft indicates that this will be no later than September 30, 2022. Once the guidance is finalized, there will be a transition period of at least one year where companies may submit via an ESG or by physical delivery to the FDA DCC.
Are there any new format or content requirements in the FDA eSTAR draft guidance?
There are no new format or content requirements in the eSTAR draft guidance, but the eSTAR template itself has several text boxes that must be filled in with summary information that is not specified in the guidance for format and content of a 510k. The information requested for the text boxes is a brief summary of non-confidential information contained in the attachments of the submission. Therefore, these boxes can information that would normally be in the overview summary documentst that are typically included at the beginning of each section of a 510k. If your overview documents do not already have this information, then you may have some additional work to do in order to complete the eSTAR templates. An example of one of these text boxes is provided below:
Another example of additional content required by the eSTAR templates is references to page numbers. Normally the FDA reviewer has to search the submission for information that is required in their regulatory review checklist. In the new templates the submitter is now asked to enter the page numbers of each attachment where specific information can be found. The following is an example of this type of request for a symbols glossary:
Are there any changes to the review timelines for a 510k in the eSTAR draft guidance?
The eSTAR draft guidance indicates that a technical screening will be completed in 15 calendar days instead of conducting a RTA screening. I believe that the technical screening is less challenging than the RTA screening, but the FDA has not released a draft of the technical screening criteria or a draft checklist. I would imagine that the intent was to streamline the process and reduce the workload of reviewers performing a technical screening, but we only have guesses regarding the substance of the technical review and so far our performance is 100% passing (i.e. 1 of 1). The next step in the 510k review process is a substantive review. Timelines for the substantive review are not even mentioned in the new draft guidance, but the FDA usually has the review clock details in Table 1 (MDUFA III performance goals) and Table 2 (MDUFA IV performance goals) of the FDA guidance specific to “Effect on FDA Review Clock and Goals.” In both tables, the goal is 60 calendar days, and our first eSTAR submission completed the substantive review in 60 days successfully. The 180-day deadline for responding to an additional information (AI) request has not changed in the eSTAR draft guidance, but our first submission is now interactive review. I believe this suggests that companies may have a higher likelihood of having an interactive review with their CDRH lead reviewer instead of being placed upon AI Hold, but we won’t have enough submissions reviewed by the FDA to be sure until the end of Q1 2022.
Register for our new webinar on the FDA eSTAR draft guidance
We hosted a live webinar on Thursday, October 21, 2021 @ Noon EDT. The webinar was approximately 37 minutes in duration. In this webinar we shared the lessons learned from our initial work with the eSTAR template. Anyone that registers for our webinar will also receive a copy of our table of contents template that we updated for use with the eSTAR templates. Unlike a 510k eCopy, an eSTAR template does not require a table of contents but we still use a table of contents to communicate the status of the 510(k) project with our clients. Finally, we reviewed the eSTAR draft guidance in detail. If you would like to receive our new eSTAR table of content template and an invitation to our live webinar, please complete the registration form below.
About the Instructor
Rob Packard is a regulatory consultant with ~25 years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510(k) submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Twitter.
How much a 510k costs is the most common question I receive from customers, and there are three parts to the cost of a 510k.
There are three parts to the 510k cost of submission:
Testing
Submission Preparation
FDA User Fees
The highest cost is testing
The testing cost is the most significant cost, but I think the average is around $100K for our clients. For devices that only consist of a software (i.e., software as a medical device or SaMD), the testing costs are less, but the cost of documenting your software validation and cybersecurity will be more extensive than the cost of preparing your 510k and the FDA user fee. The more you can do in-house, the lower the testing costs will be. Biocompatibility testing for a non-invasive device might be only $13,000, but a long-term implant can cost as much as $100,000 for implantation studies. Sterilization validation testing depends upon the method of sterilization and whether you are doing a single-lot method or a full validation. Typical costs for EO sterilization validation are around $15,000, and then you should add several thousand more for the shelf-life testing.
For devices that are powered and/or have software, you will need to perform software validation in accordance with IEC 62304 ed 1.1 (2015). There are also five FDA guidance documents that apply:
You can do all of the software validation in-house, but some firms outsource the software validation. In the long term, you need to learn this, and it pays to hire an expert in IEC 62304 to help your team learn how to document software validation if you have not done this before. Typically, software validation documentation will be between 300 and 1,000 pages long.
Electrical safety and EMC testing are often the most expensive part of the testing process for our customers. EMC testing should always be done first to ensure you can pass the immunity and emissions testing. If you must modify the device to pass the EMC testing, you must repeat any electrical safety testing. The total cost of this testing is typically $50-60K.
Performance testing is the last part of the testing process. Performance testing should be performed on sterile and aged products if your product requires sterility and claims a shelf-life. Most of the testing is benchtop testing only to demonstrate performance. This includes simulated use testing (e.g., summative usability testing), cadaver testing, and computer modeling. Benchtop performance testing typically takes tens of thousands of dollars to complete, but you might be able to do the testing in-house. If animal testing is required, this typically costs around $100K. Finally, if a human clinical study is required (i.e., ~10% of 510k submissions), you should expect to spend between $250K and $2.5 million. Some simple clinical studies (e.g., IR thermometers) cost less than $100K, but these resemble benchtop performance testing in many ways.
The second highest cost is the cost of submission preparation
Medical Device Academy has two different options for preparation consulting fees. Your first option is hourly consulting fees. The second option is a flat fee. As of July 2023, we are charging $3,500 for pre-submission preparation and $17,500 for 510k submission preparation.
The first option is to avoid the FDA altogether and submit to a third-party reviewer. We only recommend one third-party reviewer (i.e., Regulatory Technology Services), because the other companies do not have sufficient experience to have predictable review times and positive outcomes. The typical RTS third-party review cost is 6% more than the FDA Standard fee.
The second option is to submit directly to the FDA. The standard user fee for FDA review of a 510k is $21,760 for FY 2024.
The third option is to apply for small business status. For companies that have annual revenues of less than $100 million USD, the FDA will grant you small business status. For companies with small business qualifications, the FDA user fee is reduced to $5,440.
Reduce 510k cost by applying for small business status
Any medical device company with revenues of less than $100 million annually can apply, but you must apply each year. There is no application fee, but you must complete FDA Form 3602 if you are a US firm. The form must be completed for each subsidiary too. FDA Form 3602A must be completed for foreign firms, and the national tax authority must verify the accuracy of your income statement on the form to submit it to the FDA. If your national tax authority refuses to sign the form, you can justify it, but I don’t know anyone who has done this yet. The qualification review by the FDA requires 60 days. Therefore, you should apply every year in August for the next fiscal year (October 1, 2023 – September 30, 2024, is FY 2024). The form will request that you include your Organization ID #. A Dun & Bradstreet Number (DUNS #) is also required if your firm is located outside the USA. Finally, you need to attach a copy of your tax return. Therefore, you must file your tax return–even if your firm had a loss or had no revenues. You can also use R&D tax credits in the USA or Canada if you are a start-up device company developing a new device.
About the Author
Rob Packard is a regulatory consultant with 25+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Robert was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Robert’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510(k) submissions. The most favorite part of his job is training others. He can be reached via phone at 802.258.1881 or by email. You can also follow him on Google+, LinkedIn,YouTube, or Twitter.
This article helps you understand how to pass the FDA Refusal to Accept (RTA) screening process 510k submissions – updated April 2022.
What is an RTA Checklist?
The “RTA” in RTA Checklist stands for Refuse to Accept. The FDA uses this tool to determine if your 510(k) submissions will be accepted or not for a substantive review. Accepted, not approved because this is simply a verification that the required information is included in your submission. As stated in the 2022 FDA guidance document for the FDA’s Refuse to Accept Policy for 510(k)s “a minimum threshold of acceptability and should be accepted for substantive review.”(Ref.1). That does a nice job summarizing the RTA checklist. It is a tool used to help assess whether or not your submission contains the required information to continue with a more thorough review of the contents of the submission itself.
What does the Refusal to Accept (RTA) policy apply to?
The Refusal to Accept (RTA) policy applies to all 510k submissions. The RTA checklist or more checklists apply specifically to each 510(k) submission type:
Traditional 510k
Abbreviated 510k
Special 510k
There is a different RTA checklist for each submission type. The checklists are in the Refuse to Accept Policy for 510(k)s guidance document. Specifically, in the PDF document that the FDA reissued in April 2022, the checklists can be found in the following areas:
Traditional 510k – Appendix A.
Abbreviated 510k – Appendix B.
Special 510k – Appendix C.
Note that the title of the checklist refers to an ‘acceptance checklist.’ It is not called the RTA checklist until you get to the footer of the page. It is also listed as an acceptance checklist on the FDA website. The best way to think of the process is as preliminary screening by the FDA.
What does the FDA look at during the Refusal to Accept (RTA) screening process?
During the screening process, the assigned RTA screener will review the 510k submission and try to identify all of the requirements listed in the applicable RTA checklist. The person screening your submission is required to answer “yes,” “no,” or “n/a” to the questions in the checklist. This person must also enter the document and the page where the information can be found in the submission. Finally, if an element required by the refusal to accept (RTA) checklist cannot be found, then the screener adds a comment at the end of that section in the checklist. The comment will state what your deficiency is and it may even identify a guidance document that can help you address the issue. If you are missing requirements, you will receive an email from the RTA screener with the completed RTA checklist attached. We call this an “RTA Hold” letter. If your submission is not rejected, then your 510k is administratively complete, and you will receive an automated email indicating that your submission was accepted and the substantive review will begin.
Refusal to Accept (RTA) Time Frame
As stated in the guidance document, the Refusal to Accept policy includes “an early review against specific acceptance criteria and to inform the submitter within the first 15 calendar days after receipt of the submission if the submission is administratively complete, or if not, to identify the missing element(s).” (Ref. 1). If the assigned screening person is unable to complete the process within 15 calendar days, then you will receive an automated email stating that they were unable to complete the RTA checklist within 15 calendar days, and your submission is automatically moved to the substantive review stage of the 510k review process.
Taking the time to perform your gap analysis before submitting could avoid a simple error. For example, if you forget to include the signed Truthful and Accuracy Statement in your submission, it could take 15 days to be notified of that missing element. The person screening your submission could email you to provide this missing element in an interactive review to avoid placing your submission on hold. Still, they are not required to give you a chance to provide this interactively by email. If you do receive an RTA Hold letter, you might be able to correct missing elements on the same day, but the 510k review clock is automatically reset when your 510k is placed on RTA Hold. When you respond to an RTA Hold letter, there will be another 15-day refusal to accept (RTA) screening of your submission.
What do you do with the information in the comments of the RTA checklist?
The RTA checklist is the criteria that your submission is being evaluated against. Suppose your submission has deficiencies during the initial review against the RTA Checklist. In that case, the FDA will refuse to accept it, and the substantive review will not begin until those deficiencies have been corrected. Since the FDA does not hide what they are looking for or how they will evaluate your submission, use that to your advantage. Assuming that you have correctly determined the type of 510k submission you have, perform a gap analysis of your submission against the RTA checklist. Either perform these actions in-house or hire an outside consultant to do them for you, but make sure you don’t try to check your own work because you will miss something.
Scope of the FDA Refusal to Accept Guidance Document
The scope of the FDA guidance document is provided for the benefit of the FDA personnel reviewing your submission and not specifically for the 510k submitter. It also provides a loose framework for systematically and consistently reviewing submissions. This ensures all submissions receive equal, nonbiased treatment. There are some things that this guidance document does not address or alter by its own admission. One is the “substantial equivalence decision-making process once the submission has been accepted for review.” Refusing to accept (RTA) guidance also does not address FDA user fees. Other guidance documents address those issues.
What are the most common reasons for FDA refusal of your 510k submission?
Although there are dozens of reasons (43 to be exact) why the FDA could reject your submission in the 35-page RTA checklist, most of the refusals (~80%) result from a small percentage (~20%) of reasons. The most common is that your submission is poorly organized. Either you did not provide a table of contents, your submission is not organized in accordance with the sections outlined in the guidance, or the pages of your submission are not properly numbered. When trying to review a 1,200-page submission, poor organization is extremely irritating and wastes the reviewer’s time. If it were my decision, I would refuse to complete the entire checklist until you gave me a properly organized submission.
The second most common reason for refusal is submitting a device description that is inadequate. The FDA needs more detail than most companies provide for the device description because they need to understand the differences between your device and the predicate device. This includes much more than just the indications for use. Who are the intended patients and users? What is the intended environment of use? What are the materials for patient-contacting components? What is the source of power for your device? Which design features does your device include when compared to the predicate? What is the user interface for your device? Which accessory devices are needed with your device? You can even make the mistake of being inconsistent in your submission by not repeating the content in the device description in other sections of the 510k submission. It is important to duplicate certain content verbatim in other documents, such as the 510k summary, the executive summary, the substantial equivalence comparison, and the instructions for use. Paraphrasing and summarizing certain information will not work.
The third most common reason for refusal of your submission is likely related to software validation documentation. In addition to complying with the recognized IEC 62304 standard, you must also comply with the five software guidance documents published by the FDA. The FDA and 3rd-party reviewers use an 11-item checklist based on the 2005 FDA guidance document on software validation documentation. In addition, if your device has any of the following five elements, your submission must also comply with the two FDA guidance documents on cybersecurity:
Cloud communication
Network connection (active or not)
Wireless communication in any form
USB/serial ports/removable media
Software upgrades (this includes patches)
Finally, biocompatibility is the one testing section of your 510k submission that is most likely to result in refusal to accept by the FDA out of the seven sections requiring testing reports. There are several reasons why biocompatibility results in more refusals than the other six testing sections. First, the FDA requirements go above and beyond the ISO 10993-1 standard requirements. Second, the FDA requires that you submit full testing reports for biocompatibility, while you can submit summaries for other sections (e.g., sterilization validation). Third, many submitters try to provide a rationale for why testing is not required for their device. Still, the FDA has very stringent requirements for the use of a biological risk assessment or a biocompatibility certification statement in lieu of testing.
Do you have to follow the RTA checklist exactly?
You can, but you are also not bound by it. Like all guidance documents, they “contain nonbinding recommendations”. The checklist is released as part of a guidance document, so it is a guidance and not a regulatory requirement. That being said, if your submission is missing an element in the checklist, your 510k submission will be considered administratively incomplete unless you provide a clear explanation as to why the checklist element does not apply to your submission or you explain how you meet the 510k submission requirement in another way.
Medical devices vary wildly, and there is no one size fits all approach. The FDA recognizes that and includes some wiggle room that gives them some discretion in reviewing submissions. However, 100% of the 3,500+ submissions received each year are screened using the refusal to accept (RTA) checklist, and the screening person’s job is to verify that your submission meets the criteria. As it says in the guidance document:
“The purpose of the 510(k) acceptance review is to assess whether a submission is administratively complete, in that it includes all of the information necessary for FDA to conduct a substantive review. Therefore, the submission should not be accepted and should receive an RTA designation if one or more of the items noted as RTA items in the checklist are not present and no explanation is provided for the omission(s). However, during the RTA review, FDA staff has the discretion to determine whether missing checklist items are needed to ensure that the submission is administratively complete to allow the submission to be accepted. FDA staff also has the discretion to request missing checklist items interactively from submitters during the RTA review. Interaction during the RTA review is dependent on the FDA staff’s determination that outstanding issues are appropriate for interactive review and that adequate time is available for the submitter to provide supporting information and for FDA staff to assess responses. If one or more items noted as RTA items on the Acceptance Checklist are not present, FDA staff conducting the acceptance review should obtain management concurrence and notify the designated 510(k), contact person, electronically that the submission has not been accepted. “ (Ref. 1).
The portion above notes that explanations may be provided for omitted portions of the submission. So, the answer to the question is that no, you do not have to follow the RTA checklist exactly. However, if you should purposefully omit a section you should provide an explanation and your rationale justifying why the omission is appropriate for your individual device and 510(k) submission. Again, just because you have included an alternative approach or justification does not automatically mean it will be accepted. The FDA personnel who are conducting the acceptance review will judge whether or not your deviation is acceptable.
What if your 510k submission is refused?
If your submission is refused, you will be provided with a copy of the completed RTA checklist, and each of the deficiencies you must address will be highlighted. Sometimes, there will be an attachment to the checklist that has additional issues that are not in the RTA checklist, but the reviewer thinks you may need to address them later. You might also see comments that are not highlighted. These are suggestions from the reviewer that you may or may not choose to address.
There is a 180-day timeline for response to an RTA Hold letter. The response must be submitted to the CDRH Document Control Center (DCC) as an eCopy, and the response must be received within 180 days. If the response is not received within 180 days, your submission will be automatically withdrawn on the 181st day. Your response may not be piecemeal. You must address all of the issues in the RTA checklist or your submission will be placed on RTA Hold again (i.e., RTA2). If you are not sure how to organize your response, a previous blog posting and YouTube video address this topic directly.
About the Author
Matthew Walker – QMS, Risk Management, Usability | Human Factors Engineering, Cybersecurity & DFIR
Matthew brings a unique background as a former Firefighter/EMT and Rope Rescue Tech with experience in OSHA and NFPA regulations. For the better part of a decade, he has worked as a Technical/Medical Writer and Lead Auditor. He holds degrees in Fire Science and Computer Forensics and Digital Investigations, graduating Summa Cum Laude from Champlain College. Matthew is also an active member of several academic honor societies including Omicron Sigma Sigma’s Order of the Sword and Shield. His professional focus includes Human Factors Engineering, Risk Management, and Cybersecurity with a special interest in applying Digital Forensics and Incident Response (DFIR) practices to medical technology. He combines regulatory expertise with technical insige to strengthen both product safety and oranizational resiliance. He can be reached by email. You can also follow him on LinkedIn or YouTube.
A poor RTA response will cause a two-week delay, but an additional information request only gets one chance to avoid the dreaded NSE letter.
An Additional Information Request (i.e. AI Request) is typically received just before the 60th day in a 90-day 510k review, while a Refusal to Accept (RTA) Hold is typically received on the 15th day. If your response to your first RTA Hold (i.e. RTA1) is inadequate, the reviewer will issue another RTA Hold letter (i.e. RTA2) and your 510(k) review clock will be reset to 0 days. You will have another 180-days to respond to RTA2, and issues identified in an RTA Hold are usually easy to address. Most RTA Hold issues also have one or more guidance documents that are available to help you to obtain an RTA Accept letter. You can always request a submission-in-review (SIR) meeting to clarify what information the reviewer needs to address the RTA deficiencies too. If you want to learn more about responding to an RTA Hold, please read last week’s blog. The rest of this article is specific to responding to requests for additional information.
What happens after 60 days during a 510k review?
On the 60th day of the 510k review clock, or a few days prior to the 60th day, the lead reviewer must determine if they need to issue an Additional Information (AI) Request. The alternative to an AI Request is for the lead reviewer to issue a letter indicating that you have entered the Interactive Review Phase. This only happens if the reviewer believes they can make a decision regarding substantial equivalence in the next 30 days. If the decision is to issue an Interactive Review Letter, then the lead reviewer believes that only minor issues remain and there is only the need for interactive email responses between the lead reviewer and the submitter. An interactive review is the ideal outcome of the substantive review process but it rarely happens.
If you receive an Additional Information Request, what are your options?
The AI letter will indicate that you have 10 days to request a clarification meeting with the reviewer. The wording of this section of the AI letter is provided below:
“FDA is offering a teleconference within 10 calendar days from the date on this letter to address any clarification questions you may have to pertain to the deficiencies. If you are interested in a teleconference, please provide (1) proposed dates and (2) a list of your clarification questions via email at least 48 hours before the teleconference to the lead reviewer assigned to your submission. We would like to emphasize that the purpose of the meeting is to address specific clarification questions. The teleconference is not intended for the review of new information, test methods, or data; these types of questions could be better addressed via a Submission Issue Q-Submission (Q-Sub). For additional information regarding Q-Subs, please refer to the Guidance for Industry and FDA Staff on Medical Devices: Requests for Feedback and Meetings for Medical Device Submissions at https://www.fda.gov/media/114034/download.”
If you wait too long to request the teleconference, then FDA will require you to submit a formal pre-sub request or “Submission in Review” (SIR) meeting request. If you request a SIR meeting within 60 days of receiving an AI Request, the FDA will schedule a SIR meeting with you within three weeks of receiving the request–assuming resources are available. If you wait longer than 60 days to request the SIR meeting, then the FDA will default to their normal target of 60-75 days for scheduling a pre-sub meeting. For example, if you submit your SIR meeting request on day 75, and the FDA takes 75 days to schedule the meeting, you will be granted your SIR meeting at 150 days and you will only have 30 days remaining to respond to the AI Request before your submission is automatically withdrawn.
Therefore, it is important to request a clarification meeting immediately after you receive the AI Request. While you are waiting for your clarification meeting, you should immediately begin preparing any draft testing protocols that you want the FDA to provide feedback on during a SIR meeting. Then after you have the clarification meeting, you should submit your SIR meeting request and include any draft testing protocols you have prepared. This may include a statistical sampling rationale, a proposed statistical analysis method, a summative usability testing protocol, or a draft protocol for some additional benchtop performance testing. The FDA can review examples of preliminary data, a protocol, or a proposed method of analysis. The FDA cannot, however, provide a determination of substantial equivalence.
The Most Common Mistakes in Responding to an Additional Information Request
Most companies make the mistake of asking the lead review if they provide specific additional information, “Will this be sufficient to obtain 510(k) clearance?” Unfortunately, the FDA is not able to provide that answer until the company has submitted the additional information and the FDA review team has had time to review it thoroughly. This is done only when the submitter delivers an FDA eCopy to the Document Control Center at CDRH, and the review team is able to review the information. This new information is assigned a supplement number (e.g. S001), and it will typically require three weeks to review the information. Then the lead reviewer may request minor modifications to the labeling, instructions for use, or the 510k summary. This request is an interactive request, and the submitter must respond within a very short period (e.g. 48 hours), and the wording of the request may be “Please provide the above information by no later than COB tomorrow.”
FYI: “COB” means “close of business.” Wow. The FDA loves acronyms.
Best Practices in Responding to an Additional Information Request
If you receive an AI request on a Friday afternoon, 58 days after your initial submission, you should immediately request a clarification teleconference with the FDA reviewer for the following week. The only exception is if you only have minor deficiencies that you feel are completely understood. During the days leading up to the clarification teleconference, your team should send a list of clarification questions to the lead reviewer and begin drafting a response memo with a planned response to each deficiency. After the clarification meeting, you will have approximately 6-7 weeks to submit a SIR meeting request. However, you should not wait that long. Your team should make every effort to submit your SIR meeting request within 2-3 weeks. If the FDA takes 3 weeks to schedule your meeting, then you will have used approximately 6 weeks of your 26 weeks to respond to the AI Request.
In your SIR meeting request, you should always try to provide examples or sample calculations to make sure the FDA review team understands what you are proposing to submit in your supplement. For example, the FDA reviewers do not have enough time to review your entire use-related risk analysis (URRA) in a SIR meeting request. However, you can provide an example of how you plan to document a couple of use-related risks. Then you can show how these risks would translate into critical tasks. Finally, you could provide a draft summative usability testing protocol for FDA feedback. The FDA review team doesn’t have enough time available to review much more. You will only have one hour for your SIR meeting.
How to Prepare Your Response
In section “V” of the FDA guidance on deficiency responses, the FDA recommends that you restate the issue identified by the reviewer in your response. Next, your response should include one of the following:
the information or data requested, or
an explanation of why the issue is not relevant, or
alternate information with an explanation of why the information you are providing addresses the issue.
Before you respond to an AI Request, you should look up any FDA guidance documents referenced in the AI Hold letter to make sure that you address each requirement in the applicable FDA guidance document(s).
The most important technique to learn when you are responding to regulators is to organize your response in a tabular format that is numbered in exactly the same order that the request was made. Typically there will also be sub-parts to certain issues. In that case, you should duplicate the numbers and/or letters of each sub-part and segregate each sub-part in a different row of the table. Personally, I like to alternate the color of the font I use in the table to make it even more obvious which information is a restatement of the reviewer’s comment and which information is the company’s response to the AI Request.
Why you don’t get a second chance to respond to an AI Request
Once you respond to an AI Request, and the DCC receives your FDA eCopy, the FDA review clock will then resume the countdown to 90 days. In our example above, you received the AIR Request on the 58th day. The FDA must review everything you submitted and make a final substantial equivalence decision before the 83rd day because they still need to submit their recommendations to senior management in their branch. If any changes to the labeling, instructions for use, or the 510k are required, you should receive those requests several days before (i.e. 76-83 days). You can respond to interactive requests via email, and then the final SE decision will be made. If you do not respond to all of the deficiencies in the AI Request, the FDA reviewer will not have enough time to request that you address the remaining gaps and finish their review. Therefore, an incomplete AI Response will certainly result in a non-substantial equivalence (NSE) letter.
If you need to respond to an additional information request from the FDA reviewer, we can review your planned response to identify potential gaps. If you need help please use our calendly app to schedule a call with a member of our team.
About the Author
Robert Packard is a regulatory consultant with 25+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Robert was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certification. From 2009-2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Robert’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510(k) submissions. The most favorite part of his job is training others. He can be reached via phone 802.258.1881 or email. You can also follow him on Google+, LinkedIn or Twitter.