Best human factors questions?
Best human factors questions to ask the FDA during a pre-submission meeting, and what information content do you need in a 510k?
Human factors questions to ask the FDA?
The FDA did not start enforcing the requirement to apply human factors and usability engineering to medical device design until 2017 because the final version of the human factors guidance document was not released until February 3, 2016. Approximately ninety percent of the human factors testing reports submitted to the FDA in 510k pre-market submissions are deficient because the 510k submission content only includes the final summative testing report. The FDA needs a complete usability engineering file, and the human factors information needs to comply with FDA guidelines for the format and content of a 510k pre-market submission–not just IEC 62366-1:2015.

What human factors information does the FDA want?
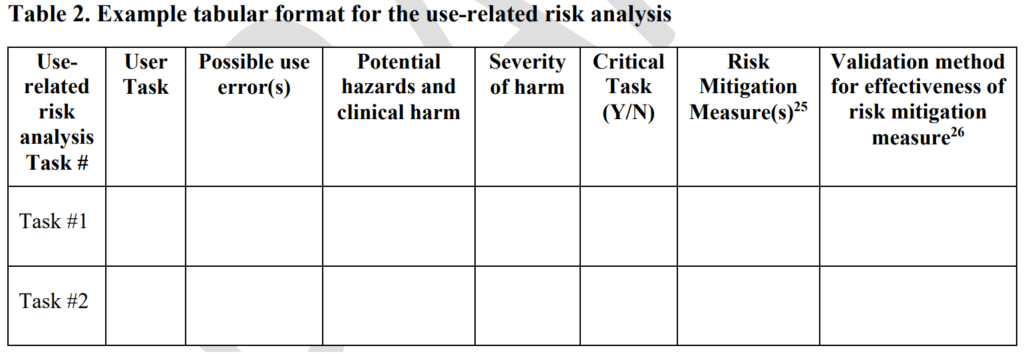
For several years, FDA submission deficiency letters indicated that you should not include the frequency of occurrence in your estimation of use-related risks. Still, the FDA never provided this information in a guidance document. On December 9, 2022, the FDA finally released a draft human factors guidance regarding the format and content of a 510k pre-market submission. The new draft guidance includes a use-related risk analysis (URRA) requirement in table 2 (copied below).
In this new draft FDA guidance, the FDA identifies three different human factors submission categories. For the first category, only a conclusion and high-level summary are needed. For the second category, a user specification is also needed. For the third category, you need a comprehensive human factors engineering report with the following elements described in Section IV of the draft FDA guidance:
Submission Category 1, 2, and 3
- Conclusion and high-level summary
Submission Category 2 and 3
- Descriptions of intended device users, uses, use environments and training
- Description of the device-user interface
- Summary of known use problems
Submission Category 3 only
- Summary of preliminary analyses and evaluations
- Use-related risk analysis to analyze hazards and risks associated with the use of the device
- Identification and description of critical tasks
- Details of validation testing of the final design
Before spending tens of thousands or hundreds of thousands of dollars on human factors testing, you want to ensure the FDA agrees with your human factors testing plan. Otherwise, you will pay for the testing twice: once for your initial submission and a second time in your response to the FDA request for additional information to address deficiencies. Testing can cost more than your electrical safety testing. The facility needs the right equipment and space for the testing; you need support personnel to set up the equipment; you need to recruit participants; you need to compensate participants; and you need device samples.
When can you ask the FDA human factors questions?
The FDA cannot provide consulting advice on a submission, and the agency will not review data during pre-submission meetings. The FDA can provide feedback on protocols, specifications, and scientific justifications. Therefore, you should submit questions to the FDA in a pre-submission when you have a draft protocol, a draft specification, or a draft justification for why a task is not critical. Pre-submissions are “non-binding.” You can change your design and approach to human factors. Therefore, don’t wait until your information is 100% finalized. Share your documentation at the draft stage during the development phase and before your design freeze. You need these answers when you are planning a study and obtaining quotes.
What are the best human factors questions to ask in a pre-sub?
In the FDA guidance for pre-submission meetings, the FDA provides suggested questions to ask:
- Does the Agency have comments on our proposed human factors engineering process?
- Is the attached use-related risk analysis plan adequate? Does the Agency agree that we have identified all the critical tasks?
- Does the Agency agree with our proposed test participant recruitment plan for the human factors validation testing?
The above examples are only suggestions, but the best approach is to provide a brief example of what the human factors information will look like and ask the FDA to comment on the examples. The FDA does not have time to review data during a pre-sub meeting, but the FDA can review a few rows extracted from your URRA and comment on your proposed approach to the human factors process.
Human factors questions that are not appropriate
The FDA pre-submission guidance cautions you only to ask 3-4 questions for each meeting request because the FDA has difficulty answering more questions in a 60-minute teleconference. Therefore, you should not ask questions already answered in the FDA guidance. The new draft guidance includes examples of when a device modification can leverage existing human factors information and when new information is needed to support a premarket submission. Instead of asking a question specific to leveraging existing human factors information, provide your rationale for leveraging existing data and ask if the FDA has any concerns with your overall approach to human factors.
Recommended human factors action items
Create a procedure for your human factors process that includes detailed instructions for creating the information required in a usability engineering report and templates for each document.
Best human factors questions? Read More »