90% of usability testing submitted to the FDA is unacceptable and the root cause is simply a failure to understand the human factors process.
Human factors process inadequate?
If you submitted no usability testing to the FDA in your 510(k) submission, it would be obvious why the FDA reviewer identified usability as a major deficiency. However, you spent tens of thousands of dollars on usability testing that delayed the 510(k) submission by six months. Despite all of the time and money your company invested in the human factors process, it appears that you need to start over and repeat the entire process again. The CEO is furious, and he wants you to show him where in the 49-page FDA guidance it says that you have to do things differently.
Benefits from the human factors process
- Use errors result in serious injuries and death
- Easy-to-use products sell
- You will prevent delays in regulatory approval
Why was your rationale for no usability testing rejected?
Unlike CE Marking technical files, the FDA does not require a usability engineering file for all products. Instead, the FDA determines if usability testing is required based on a comparison of your device’s user interface and a competitor’s user interface (i.e. predicate device user interface). If the user interface is identical, then usability testing may not be required. Instead, your company should be able to write a rationale for not doing usability testing based on equivalence with the predicate device. If there are differences in your user interface, you will need to provide use-related risk analysis (URRA), identify critical tasks, implement risk controls, and provide verification testing to demonstrate the effectiveness of the risk controls. Even if your device is “easier to use” or “simpler”, you still need to provide the documentation to support this claim in your submission. The FDA also does not allow comparative claims in your marketing for 510(k) cleared devices. Comparative claims require the support of clinical data.
What is the 10-step human factors process?
- Define human factors for your device or IVD
- Identify use errors
- Conduct a use-related risk analysis (URRA)
- Perform a critical task analysis
- Conduct a risk control option analysis
- Conduct formative usability testing
- Implement risk controls
- Conduct summative usability testing
- Prepare HFE/UE documentation
- Collect post-market surveillance data specific to use errors
There is a YouTube video describing these 10 steps at the bottom of this blog posting.
Usability Engineering & Human Factors Training Webinars
- Human Factors Training Series – $950
- Formative Usability Testing Webinar & Template Bundle – $79
- Use Error and Abnormal Use Decision Tree Training – $79
- Use Specification Template & Webinar Bundle – $79
- Known Use Errors Search Webinar – $129
- Task Analysis Template & Webinar Bundle – $129
- Use-Related Risk Analysis (URRA) Template & Webinar Bundle – $299
- Participant Screening & Data Collection Forms – $79
- Summative Usability Testing Protocol & Webinar Bundle – $199
- Summative Usability Testing Report – Live-streaming Free Webinar
Why is formative testing needed?
- Observational study to identify unforeseen use errors
- Observational study to evaluate risk control options
- What are the other types of studies?
- Development of indications for use
- Development of training materials
Why is the human factors process crazy expensive to outsource?
- Human factors consultants need time to learn about your device
- Consultants are more conservative because they cannot afford to fail
- Justifying your choice of risk controls is difficult because you started too late
- Your instructions for use (IFU) are inadequate
- Consultants need to explain the human factors process to you
- Recruiting subjects is marketing (which may not be their expertise)
- You are paying for infrastructure (specialized testing facilities)
- This is a team effort that requires many consulting hours collectively
Why was your Usability Engineering File refused?
- Your company provided an application failure modes and effects analysis (aFMEA) to support your justification that residual risks are acceptable. The FDA guidance suggests using risk analysis tools such as an FMEA or fault-tree analysis, but deficiency letters from FDA reviewers recommend a use-related risk analysis (URRA) format that is totally different.
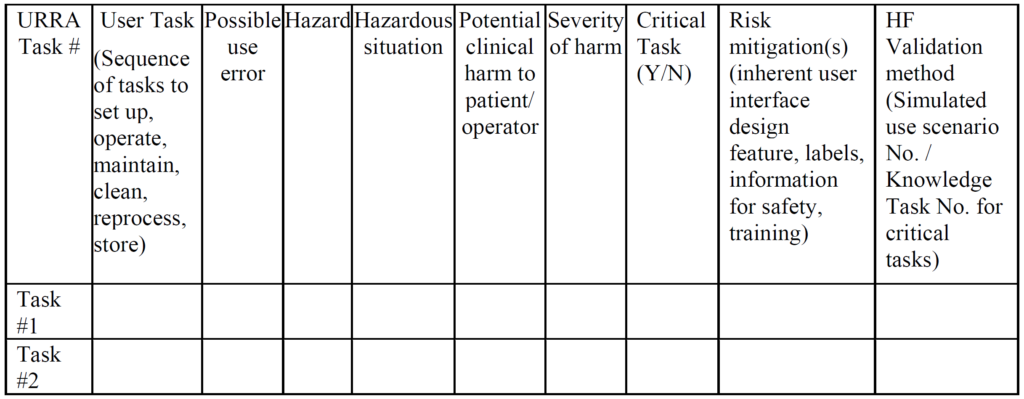
Example of a URRA Table provided by the FDA for the Human Factors Process The primary problem with using an FMEA or Fault-Tree risk analysis tool is that these tools involve the estimation of the severity of harm and the probability of occurrence of harm, while the FDA does not feel it is appropriate to estimate the probability of occurrence of harm. Instead, the FDA instructs companies to assume that use errors will occur and to implement risk controls to mitigate those risks (see URRA example above). Although “mitigation” is unlikely, and use risks will only be reduced, this is the approach the FDA wants companies to use. In addition, the FDA expects your company to provide traceability of risk control implementation to each use-related risk you identified and the FDA expects documentation of verification testing (i.e. usability testing) that shows your risk controls are effective. Finally, the FDA (and ISO 14971, Clause 10) expects you to collect and perform a trend analysis of use errors. Any use errors that are reported should be evaluated for the need to implement additional corrective actions to prevent future use errors. Blaming “user error” is not an acceptable approach.
- You provided risk analysis and human factors testing in your 510(k) submission, but the FDA reviewer said you need to identify critical tasks and provide traceability to each critical task in your summative validation report. – Critical tasks are specifically mentioned in section 3.2 of the FDA guidance on applying human factors and usability engineering–and a total of 49 times throughout the guidance. However, “critical tasks” are not mentioned even once in ISO 14971:2019 or ISO/TR 24971:2020. The term “critical tasks” is not even found in IEC 62366-1:2015. There is mention of “tasks”, and “task” is a formal definition (i.e. Definition 3.14, “Task – one or more USER interactions with a MEDICAL DEVICE to achieve a desired result”). Therefore, companies that are familiar with the ISO Standards and CE Marking process frequently need training on the FDA requirements for the human factors process. After receiving training, then your company will be prepared to modify your usability engineering file documentation to comply with the FDA requirements for human factors.
- You completed a summative validation protocol, but the FDA disagrees with your definition of user groups. – Each user has a different level of experience, training, and competency. Therefore, if you define the intended user population too broadly (e.g. healthcare practitioners), the FDA may not accept your summative usability testing. This is the reason that the human factors process begins with defining the human factors for your IVD or device. Radiologists, for example, have the following training pathway:
- graduate from medical school;
- complete an internship;
- pass state licensing exam;
- complete a residency in radiology;
- become board certified; and
- complete an optional fellowship.
Therefore, if you are developing imaging software, you need to make sure your user group includes radiologists that cover the entire range of competencies. In addition, most radiology images are taken by radiology technicians and then reviewed by the radiologist. Therefore, radiology technicians should be considered a completely different user group due to the differences in experience, training, and competency when compared to a radiologist. This simple example doubles the number of users needed because you have two user groups instead of one.
- You evaluated 15 users, but the FDA reviewer is asking you to evaluate a larger number of users based on a special controls guidance document. – The FDA guidance on human factors testing specifies a minimum of 15 users for each user group–not a minimum of 15 users. Therefore, for a device that is for Rx-only and OTC use, you will have at least two user groups that need to be evaluated independently. In addition, some devices have special controls guidance documents that specify usability testing requirements. For example, an OTC blood glucose meter must pass a 350-person lay-user study. Covid-19 self-tests are expected to pass a 30-person lay-user study as another example.
- Your usability study was conducted in Australia, but the FDA insists that your usability study must be repeated in the USA. – Most people think of language being the primary difference between two countries, and therefore the author of a study protocol may not perceive any difference between the USA and Australia, Ireland, Canada, or the UK. However, this lack of ability to identify differences between cultural norms shows our own ignorance of cultural differences. International travelers learn quickly about the differences in the interface used for electrical outlets between the USA and other countries. There are also more subtle differences between cultures, such as in which direction do you toggle a light switch to turn on a light, up or down? For devices that are used in a hospital environment, it is critical to understand how your device will interact with other devices and how different hospital protocols might impact human factors.
- The FDA reviewer indicated that your usability engineering file does not assess the ability of laypersons to self-select whether your OTC device is appropriate for them. – Devices and IVD devices may have contraindications or indications for use that are specific to an intended patient population or intended user population. In these cases, the user of the device or IVD needs to be able to “self-select” as included or excluded from use. The ability to self-select should be assessed as part of any OTC usability study. The ability to identify suitable and unsuitable patients for treatment is also a common criterion for a usability study involving prescription devices where a physician is the subject of the study.
- The FDA reviewer indicated that you did not provide raw data collected by the study moderator. – Data collected during a human factors study is usually subjective in nature, and the FDA may want to conduct its own review and analysis of your data. Therefore, you cannot provide only a testing report that summarizes the results of your study. You must also provide the raw data for the study. It is permitted to provide the data in a tabular format that has been transcribed from paper case report forms or was recorded electronically. You should also consider scanning any paper forms for permanent retention or retaining the paper forms in case there is any question of accuracy in the transcription of the data collected. Finally, it is best practice to record videos of the study participants performing each task and answering interview questions. This will help in filling any gaps in the notes recorded by the moderator, and the recording provides additional objective evidence of the study results.
- The FDA reviewer indicated that your study is not valid, because the training provided by moderators was not scripted and training decay was not considered in the design of the study. – Summative usability testing requires that users complete all of the critical tasks identified in your critical task analysis without assistance. It is permitted to provide training to the user prior to conducting the study if the device or IVD is for prescription use and healthcare practitioners are responsible for providing instruction to the user. However, any training provided must be scripted in advance and approved as part of the summative usability testing protocol. This ensures that every subject in the study receives consistent training. Unfortunately, the FDA may still not be satisfied with the design of your study if you do not allow sufficient time to pass between the time that training is provided to the user and when the subject uses the device or IVD for the first time. In general, one hour is the minimum amount of time that should pass between providing user training and when the device or IVD is used for the first time. This is referred to as “training decay” and the duration of time between your scripted training and the user performing critical tasks for the first time should be specified in your summative usability protocol. One solution to address both issues is to provide a video of the instructions to each subject 24-hours in advance of participation in the study.