This article explains how you should be integrating usability testing into your design control process–especially formative usability testing.
Why you should be integrating usability testing into the design
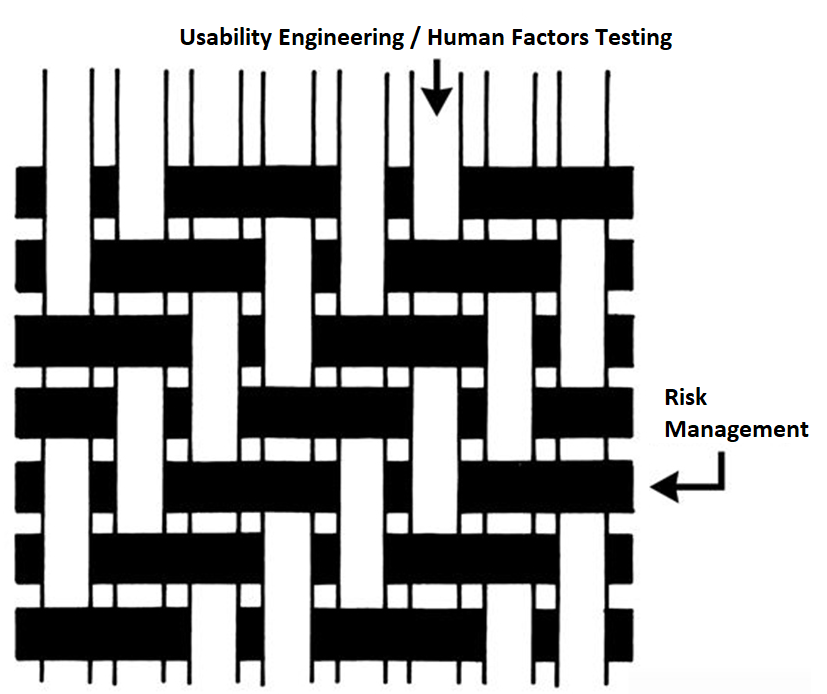
We recently recorded an updated usability webinar and released a usability procedure (SYS-048) with help from Research Collective–a firm specializing in human factors testing. After listening carefully to the webinar, and reading through the new usability procedure, I felt we needed to update our combined design/risk management plan to specify formative testing during phase 3 and summative (validation) testing during phase 4 of the design process. This is necessary to ensure your usability testing is interwoven with your risk management process. Integrating usability testing into all phases of your design process is critical–especially design planning (phase 1), feasibility (phase 2), and development (phase 3).

Integrating usability testing into your design plan helps identify issues earlier
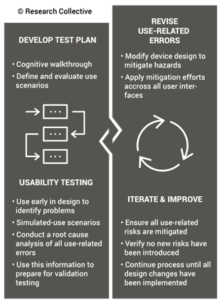
During the usability training webinar, Research Collective provided a diagram showing the various steps in the usability engineering process. The first five steps should be included in Phases 1 and 2 of your design process. Phase 1 of the design process is planning. In that phase, you should identify all of the usability engineering tasks that need to be performed during the design process and estimate when each activity will be performed. The first of these usability activities is the identification of usability factors related to your device. Identifying usability factors is performed during Phase 2 of your design process before hazard identification.
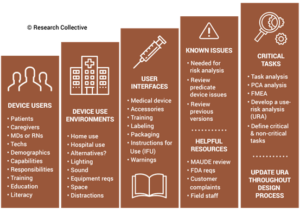
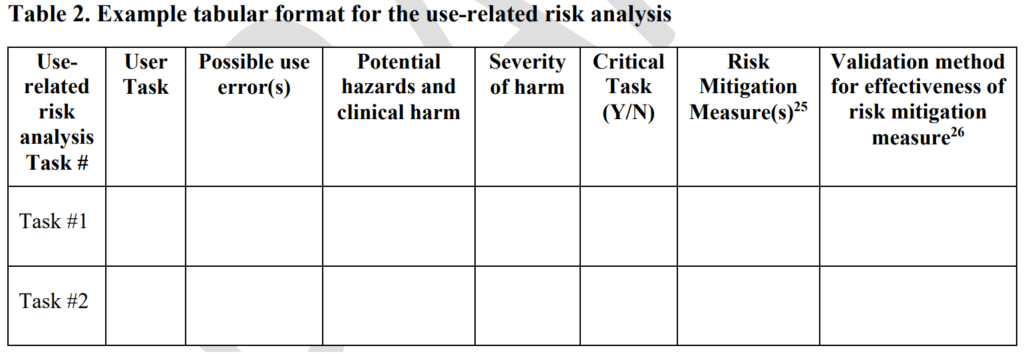
Before performing hazard identification, which should include identifying potential use errors, you need to identify five key usability elements associated with your device:
- prospective device users during all stages of use must be defined
- use environments must be identified
- user interfaces must be identified
- known use errors with similar devices and previous generations of your device must be researched
- critical tasks must be described in detail and analyzed for potential use errors
Defining users must include the following characteristics: physical condition, education, literacy, dexterity, experience, etc. Use environment considerations may consist of low lighting, extreme temperatures or humidity, or excessive uncontrolled motion (e.g., ambulatory devices). User interfaces may include keyboards, knobs, buttons, switches, remote controllers, or even a touch screen display.
Often the best reason for developing a new device is to address an everyday use error that is inherent to the design of your current device model or a competitor’s product. Therefore, a thorough review of adverse event databases and literature searches for potential use errors is an important task to perform before hazard identification. This review of adverse event data and literature searches of clinical literature are key elements of performing post-market surveillance, and now ISO 13485:2016 requires that post-market surveillance shall be an input to your design process.
Finally, the step-by-step process of using your device should be analyzed carefully to identify each critical user task. User tasks are defined as “critical” for “a user task which, if performed incorrectly or not performed at all, would or could cause serious harm to the patient or user, where harm is defined to include compromised medical care.” Not every task is critical, all critical tasks must be identified, and ultimately you need to verify that each critical task is performed correctly during your summative (validation) usability testing.
Evaluating Risk Control Options – Formative Usability Testing in Phase 3 (Development)
Once your design team has conducted hazard identification and identified your design inputs (i.e., design phase 2), you will begin to evaluate risks and compare various risk control options. Risk control option analysis requires testing multiple prototype versions to assess which design has the optimum benefit/risk ratio. This is an iterative process that involves screening tests. For any use risks you identify, formative usability testing should be performed. Sometimes the risk controls you implement will create new use errors or new risks of other types. In this case, you must compare the risks before implementing a risk control with risks created by the risk control.
Ideally, each design iteration will reduce the risks further until all risks have been eliminated. The international risk management standard (ISO 14971) states that risks shall be reduced as low as reasonably practicable (ALARP). However, the European medical devices regulations require risks to be reduced as far as possible, considering the state-of-the-art. For example, all small-bore connectors in the USA are now required to have unique connectors that are incompatible with IV tubing Luer lock connections to prevent potential use errors. That requirement is considered “state-of-the-art.” If your device is marketed in both the USA and Europe, you will need to reduce errors as far as possible–before writing warnings and precautions in your instructions for use.
Reaching the point where use errors cannot be reduced any further may require many design iterations, and each iteration should be subsequently evaluated with formative usability testing. Formative testing can be performed with prototypes, rather than production equivalents, but the formative testing conditions should also address factors such as the use environment and users with different levels of education and/or experience. Ultimately, if the formative testing is done well, summative (validation) testing will be a formality.
Risk Control Effectiveness During Phase 4 – Summative Usability Testing during Verification
Once your team freezes the design, you will need to conduct verification testing. This includes integrating usability testing into the verification testing process. Summative (validation) testing must be performed once your design is “frozen.” If you are developing an electrical medical device, then you will need to provide evidence of usability testing as part of your documentation for submission to an electrical safety testing lab for IEC 60601-1 testing. There is a collateral standard for usability (i.e., IEC 60601-1-6). For software as a medical device (SaMD), you will also be expected to conduct usability testing to demonstrate that the user interface does not create any user errors.

When you conduct summative (validation) testing, it is critical to make sure that you are using samples that are production equivalents rather than prototypes. Also, it is crucial to have your instructions for use (IFU) finalized. Any residual risks for use errors should be identified in the precautions section of your IFU, and the use of video is encouraged as a training aid to ensure use errors are identified, and the user understands any potential harm. When the summative testing is performed, there should be no deviations and no use errors. Inadequate identification of usability factors during Phase 2, or inadequate formative testing during Phase 3, is usually the root cause of failed summative testing. If your team prepared sufficiently in Phase 2 and 3, the Phase 4 results would be unsurprisingly successful.
Additional Training Resources for Usability Engineering
The following additional training resources for usability engineering may be helpful to you:

 Matthew Walker – QMS, Risk Management, Usability Testing, Cybersecurity
Matthew Walker – QMS, Risk Management, Usability Testing, Cybersecurity