This is an overview of the updated 510k electronic submission guidance document that the FDA released on October 2, 2023.
What’s included in the 510k electronic submission guidance?
As with any FDA guidance, there is an introduction and background regarding the reason for the updated guidance document (i.e., eSTAR guidance). At the very beginning of the document (i.e., page 3) the reference to the RTA Guidance was deleted, because there is no longer an RTA screening process with the implementation of the FDA eSTAR templates. The updated guidance explains on page 6 that “The CDRH Portal will automatically verify that the eSTAR is complete, and therefore we do not expect to receive incomplete 510(k) eSTARs.” In the scope section, the FDA specifies that this document is specific to 510k submissions using the eSTAR template. The document also explains that CBER conducted a pilot with the eSTAR template in June 2022 and now the FDA eSTAR template must be used in conjunction with the CDRH Portal for submission of a 510k to CBER. The FDA has plans to release a similar De Novo submission guidance for using the eSTAR template, but this has not happened in the year since the FDA announced the intention to do so. In the “Significant Terminology” section of the guidance (i.e., Section IV), the FDA provides definitions for each of the different types of submissions: eCopy, eSubmitter, etc. In the “Current Electronic Submission Template Structure, Format, and Use” section of the guidance (i.e., Section V), the FDA modified the term used for the company that is applying for 510k clearance from “Submitter” to “Applicant,” because sometimes a regulatory consultant or 3rd party reviewer is submitting the 510k on behalf of the applicant. On page 12 of the updated guidance, the FDA added “Withdrawal requests” to the list of 510k submissions/information that is exempt from the 510k electronic submission requirements. In the next to last section of the electronic submission guidance, the FDA provides a table outlining all of the sections of the new eSTAR template. The table is reproduced later in this article. If you are interested in a tutorial on completing each section outlined in the table, we recommend purchasing Medical Device Academy’s 510(k) Course. The last section of the eSTAR guidance indicates the timing for compliance with the updated guidance (i.e., October 1, 2023).

What is the deadline for compliance with the guidance?
The deadline has now passed. The new eSTAR template must be used for all 510k and De Novo submissions as of October 1, 2023. You must upload the new FDA eSTAR submissions using the CDRH Portal. You will need to request an account using a registration hyperlink.
What’s missing from this 510k submission guidance?
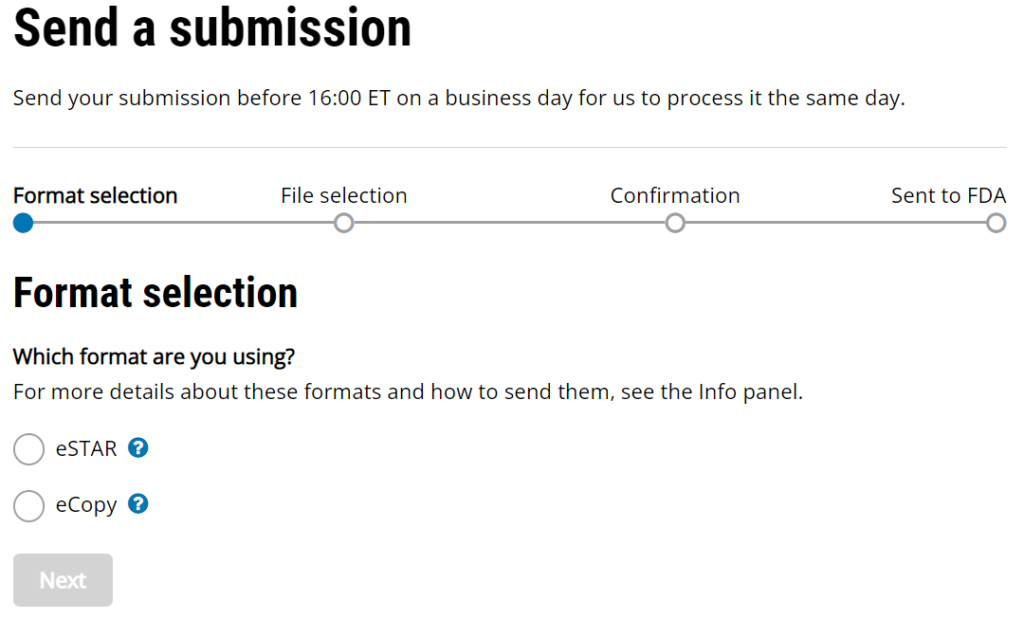
The updated 510k electronic submission guidance does not provide information regarding the receipt date for electronic submissions made through the new customer collaboration portal (CCP) created by CDRH. The image below is a screen capture of the current CCP upload webpage. It includes the following statement, “Send your submission before 16:00 ET on a business day for us to process it the same day.” This statement was added sometime in August or September, but the FDA has not released a detailed explanation. This statement makes it clear that the FDA is not promising to process a submission the “same day” if the submission is received after 4:00 p.m. ET. However, “processed” does not have the same meaning as “receipt date.”
Another element missing from this updated guidance is a reference to human factors documentation. For any devices that have a user interface that is different from the predicate device, and for software devices, the FDA requires documentation of your human factors process to make sure that differences in the user interface do not result in new or different risks when compared to the predicate device. The 2016 FDA guidance for human factors has not been updated, but FDA reviewers continue to issue deficiencies related to the objective evidence provided in a 510k for human factors validation.
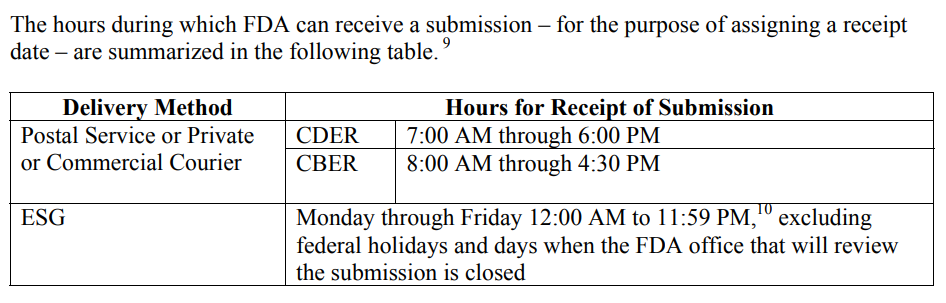
The FDA must be consistent in the wording for “Hours for Receipt of Submission” because this affects submissions at the end of the fiscal year, but it also affects any submissions with a deadline for response to an RTA Hold, AI Response, and IDE submissions. The CDER and CBER divisions of the FDA address the need for defining the date of receipt in a guidance document specific to this topic, “Providing Regulatory Submissions in Electronic Format–Receipt Date.” Below is a screen capture copied from page 4 of the guidance.
Another element missing from this new guidance is a reference to human factors documentation. For any devices that have a user interface that is different from the predicate device, and for software devices, the FDA requires documentation of your human factors process to make sure that differences in the user interface do not result in new or different risks when compared to the predicate device. The 2016 FDA guidance for human factors has not been updated, but FDA reviewers continue to issue deficiencies related to the objective evidence provided in a 510k for human factors validation.
What are the new sections for a 510k submission?
In 2019, the FDA released a guidance document on the “Format of Traditional and Abbreviated 510(k)s.” That guidance outlines the 20 sections of a traditional 510k submission that have been used for decades. However, the new 510k electronic submission guidance has no numbering for the sections of the eSTAR template, and there are 22 sections instead of 20 sections. Several of the new sections are elements of the current FDA submission cover sheet (i.e., FDA Form 3514), and some sections exist in the 2019 guidance that were eliminated, such as: “Class III Summary and Certification.” Therefore, Medical Device Academy is recreating 100% of our 510k training webinars to explain how our 510k templates are used with the 510k eSTAR template and how to fill in the PDF form. To prevent confusion between the two formats, we are using letters for each section in the eSTAR template instead of numbers (i.e., A-V instead of 1-20). Table 1 from the new eSTAR guidance is reproduced below for your information.
| Information Requested | Description | |
| A | Submission Type | Identification of key information that may be useful to FDA in the initial processing and review of the 510(k) submission, including content from current Form FDA 3514, Section A. |
| B | Cover Letter / Letters of Reference | Attach a cover letter and any documents that refer to other submissions. |
| C | Submitter Information | Information on submitter and correspondent, if applicable, consistent with content from current Form FDA 3514, Sections B and C. |
| D | Pre-Submission Correspondence & Previous Regulator Interaction | Information on prior submissions for the same device included in the current submission, such as submission numbers for a prior not substantially equivalent (NSE) determination, prior deleted or withdrawn 510(k), Q-Submission, Investigational Device Exemption (IDE) application, premarket approval (PMA) application, humanitarian device exemption (HDE) application, or De Novo classification request. |
| E | Consensus Standards | Identification of voluntary consensus standard(s) used, if applicable. This includes both FDA-recognized and nonrecognized consensus standards. |
| F | Device Description | Identification of listing number if listed with FDA.Descriptive information for the device, including a description of the technological characteristics of the device including materials, design, energy source, and other device features, as defined in section 513(i)(1)(B) of the FD&C Act and 21 CFR 807.100(b)(2)(ii)(A). Descriptive information also includes a description of the principle of operation for achieving the intended effect and the proposed conditions of use, such as surgical technique for implants; anatomical location of use; user interface; how the device interacts with other devices; and/or how the device interacts with the patient.Information on whether the device is intended to be marketed with accessories.Identification of any applicable device-specific guidance document(s) or special controls for the device type as provided in a special controls document (or alternative measures identified that provide at least an equivalent assurance of safety and effectiveness) or in a device-specific classification regulation, and/or performance standards. See “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].” |
| G | Proposed Indications for Use (Form FDA 3881) | Identification of the proposed indications for use of the device. The term indications for use, as defined in 21 CFR 814.20(b)(3)(i), describes the disease or condition the device will diagnose, treat, prevent, cure, or mitigate, including a description of the patient population for which the device is intended. |
| H | Classification | Identification of the classification regulation number that seems most appropriate for the subject device, as applicable. |
| I | Predicates and Substantial Equivalence | Identification of a predicate device (e.g., 510(k) number, De Novo number, reclassified PMA number, classification regulation reference, if exempt and limitations to exemption are exceeded, or statement that the predicate is a preamendments device).The submission should include a comparison of the predicate and subject device and a discussion why any differences between the subject and predicate do not impact safety and effectiveness [see section 513(i)(1)(A) of the FD&C Act and 21 CFR 807.87(f)]. A reference device should also be included in the discussion, if applicable. See “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].” |
| J | Design/Special Controls, Risks to Health, and Mitigation Measures | Applicable to Special 510(k) submissions only.Identification of the device changes and the risk analysis method(s) used to assess the impact of the change(s) on the device and the results of the analysis.Risk control measures to mitigate identified risks (e.g., labeling, verification). See “The Special 510(k) Program.” |
| K | Labeling | Submission of proposed labeling in sufficient detail to satisfy the requirements of 21 CFR 807.87(e). Generally, if the device is an in vitro diagnostic device, the labeling must also satisfy the requirements of 21 CFR 809.10. Additionally, the term “labeling” generally includes the device label, instructions for use, and any patient labeling. See “Guidance on Medical Device Patient Labeling.” |
| L | Reprocessing | Information for assessing the reprocessing validation and labeling, if applicable. See “Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling.” |
| M | Sterility | Information on sterility and validation methods, if applicable. See “Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile.” |
| N | Shelf Life | Summary of methods used to establish that device performance is maintained for the entirety of the proposed shelf-life (e.g., mechanical properties, coating integrity, pH, osmolality), if applicable. |
| O | Biocompatibility | Information on the biocompatibility assessment of patient contacting materials, if applicable. See “Use of International Standard ISO 10993-1, ‘Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process.’” |
| P | Software/Firmware | Submission of applicable software documentation, if applicable. See “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices.” |
| Q | Cybersecurity/Interoperability | Submission of applicable information regarding the assessment of cybersecurity, if applicable. See “Content for Premarket Submissions for Management of Cybersecurity in Medical Devices” and “Design Considerations and Premarket Submission Recommendations for Interoperable Medical Devices.” |
| R | Electromagnetic Compatibility (EMC), Electrical, Mechanical, Wireless and Thermal Safety | Submission of the EMC, Electrical, Mechanical, Wireless and Thermal Safety testing for your device or summarize why testing is not needed. See “Electromagnetic Compatibility (EMC) of Medical Devices” and “Radio Frequency Wireless Technology in Medical Devices.” |
| S | Performance Testing | For non-in vitro diagnostic devices: Provide information on the non-clinical and clinical test reports submitted, referenced, or relied on in the 510(k) for a determination of substantial equivalence. See “Recommended Content and Format of NonClinical Bench Performance Testing Information in Premarket Submissions.”For in vitro diagnostic devices: Provide analytical performance, comparison studies, reference range/expected values, and clinical study information. |
| T | References | Inclusion of any literature references, if applicable. |
| U | Administrative Documentation | Inclusion of additional administrative forms applicable to the submission, including but not limited to a general summary of submission/executive summary (recommended), a Truthful and Accuracy Statement, and a 510(k) Summary or statement. |
| V | Amendment/Additional Information (AI) response | Inclusion of responses to Additional Information requests. |
Important information in the eSTAR guidance
In Table 1 above, there are 14 hyperlinks to various FDA guidance documents. These links are extremely helpful when you have questions about a specific question. Unfortunately, the 510k electronic submission guidance document will quickly become out-of-date as guidance documents are updated and made obsolete. In particular, one of the A-list final guidance documents that was planned for FY 2023 was the FDA cybersecurity guidance. The updated cybersecurity guidance was finally released last week.
Any updated info or guidance on where/how to document the human factors and usability documentation within the eSTAR?
That’s a great question. In the Performance testing section of the eSTAR template, the first question you are asked is: Was Bench Testing used in order to support this submission?
If you answer “yes” with the dropdown menu, then the eSTAR template will create a Bench Testing subsection. In that subsection there is a button for “Add Attachment.” That is where you attach the human factors and usability documentation within the eSTAR. The help button on the right provides you with a JavaScript Window that lists all of the different types of benchtop testing that you might be submitting. The last item in the list is:
“Usability/Human Factors: Studies specifically assessing the instructions and/or device design in terms of impact of human behaviour, abilities, and other characteristics on the ability of the device to perform as intended should be included here.”
The eSTAR version I’m quoting is nIVD v2.2. There is also a new draft guidance for the premarket submission documentation requirements for human factors. That gives you a good list of what the FDA wants in this section for attachments. When that guidance is finalized, I expect the FDA might expand the benchtop testing section to include a bunch of attachments much like you find in the software section. But that could be a year from now.
My detailed tutorial on how to add this information to the eSTAR will be covered in our March 16, 2023 live webinar. You can participate in that training if you purchase our 510(k) Course.
Thanks for the response- the IVD eSTAR is on version 1 which has none of these sections for non-clinical bench testing or human factors or usability. The first question in the Performance testing section is regarding Analytical Performance, and based on my previous answers for IVD software product, jumps to clinical studies, then performance testing summary.
You’re very welcome. I’m glad it was helpful to you.
So to follow up- any direction for the IVD eSTAR?
I don’t see anywhere in the eSTAR IVD v1.5 template that human factors is specifically mentioned either. I would expect it to belong in the performance testing section under the following question?
“Do you have other Clinical Supportive Data to include in this submission?”
This section allows you to attach multiple documents and explain the purpose of the documents in a text box above the attachments. This is what I would do for a device like a glucose test meter that requires human factors testing as part of the Special Controls.
I submitted this question to Lili Duan at the FDA, and she confirmed that this is correct…
“You are correct, the Human Factors / Usability information is attached to the Performance Testing à Clinical Studies à ‘Do you have other Clinical Supportive Data to include in this submission?’ question. Please note you can continue attaching additional bench tests, etc. by clicking on Add Attachment button and selecting files. However, please ensure that you provide a brief explanation of each attachment and what area it covers.”
Thanks Rob, for the update. So even though this is NON-clinical bench performance and usability (for SaMD, IVD) it should go in the Clinical Studies section?
Hi Rob, thank you so much for the videos, very informative. I quickly wanted to find out. What format will FDA except for 510K exempt as the eSTAR is mainly for premarket and De Novo.
There is no submission to the FDA for 510(k) exempt products at all. You may be required to have a Design History File (DHF) for Class 2 devices, but that DHF will be sampled during the routine inspections and there is no required format/content.