It’s a common misconception that FDA De Novo content is very different from FDA 510k submission content, but is that true?
What do you think the De Novo content differences are?
Most people think the difference between a 510k and a De Novo is time and money. That conclusion is based upon a very important assumption: a 510k will not require clinical data, and a De Novo will require clinical data. That assumption is not always correct. 10-15% of 510k submissions include clinical data to support the performance claims, and last year our team submitted three De Novo submissions that did not include any clinical data. So what are the differences between a 510k and a De Novo content?

We use the same FDA eSTAR template for both types of FDA submissions, and on the first page of the eSTAR template, we identify if the submission is a 510k or De Novo. If we select De Novo, the eSTAR will be pre-populated with six unique De Novo content requirements covering four (4) different areas that are not found in a 510k. The six unique content requirements are:
- Recommending a classification, providing a justification for that classification, and explaining what efforts were taken to identify a suitable 510k product code
- Description of existing alternative practices or procedures used in diagnosing, treating, preventing, curing, or mitigating the disease or condition for which the IVD or device is intended
- A risk mitigation table must be provided
- Providing a written benefit/risk analysis starting with the clinical benefits of your device
- Efforts to identify a potential predicate (including identifying alternative practices, procedures, or even drugs)
- Recommendations for FDA special controls for your new product code based upon the risks to health and the mitigation measures for each risk
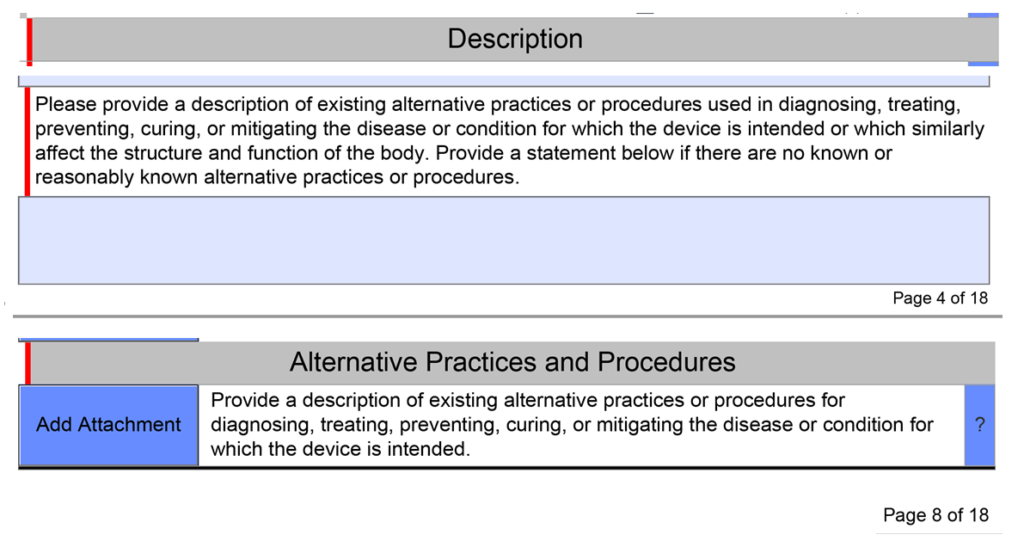
What alternative practices and procedures are currently available?
The unique De Novo content requirement is to provide a description of alternative practices and procedures for treatment or diagnosis of the same indications that you are proposing for your subject device. This is a subsection of the device description section in the FDA eSTAR template. Your should description should include other 510k-cleared products, drugs, and even products that have similar indications but are not identical. The description of alternative practices and procedures must also be attached as a document in the section for benefits, risks, and mitigation measures. To maintain consistency throughout your submission, you should create the document for attachment first and copy and paste the content into the text box at the end of the device description section.
You need to recommend a classification in your De Novo
The unique De Novo content requirement is found in a section titled “Classification.” There is a shorter classification section included in 510k submissions, but the 510k version only has four cells. The first three are populated by selecting one of the options from a dropdown menu, and the fourth cell is only used if your subject device includes other product classification codes.
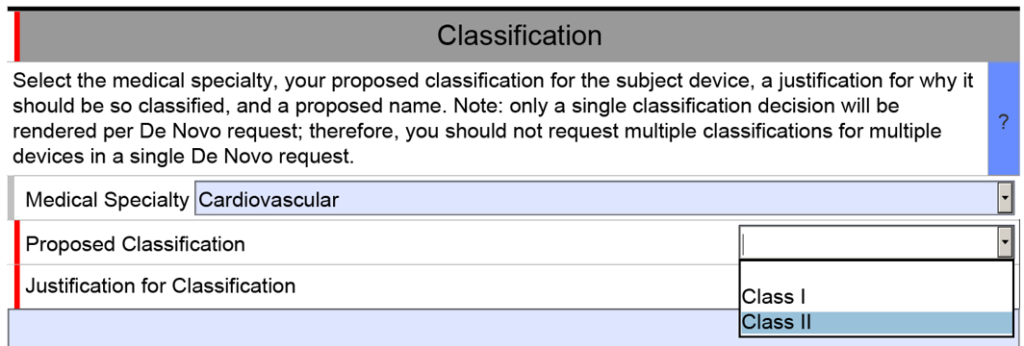
The De Novo version of the eSTAR is identical for the first row of the classification section, but then you must select a proposed product classification (i.e., Class 1 or Class 2) in accordance with FDA Classification Procedures (i.e., 21 CFR 860). The third cell is a text box for you to enter your justification for the proposed classification. Next, the FDA requires you to enter a proposed classification name. Finally, at the end of the classification section, the FDA requires that you provide a classification summary or reference to a previous NSE 510k submission.
A benefit-risk analysis is required in the De Novo content
For new devices, the FDA uses a benefit-risk analysis to decide if a device should be authorized for marketing in the USA. This process includes humanitarian device exemptions, De Novo applications, and Premarket Approval submissions. The FDA has a guidance document that provides guidance for FDA reviewers and the industry. The most important aspect is, to begin with, the benefits of the device and to provide a quantitative comparison of benefits and risks. Many De Novo submissions have been rejected because the submitter did not provide objective evidence of clinical benefits for the subject device.

The FDA guidance documents are helpful for creating a benefit-risk analysis, but you can also find information in the ISO/TIR 24971:2020–the guidance for the application of ISO 14971:2019. Our company also includes a template for a benefit/risk analysis as part of our risk management procedure (i.e., SYS-010).
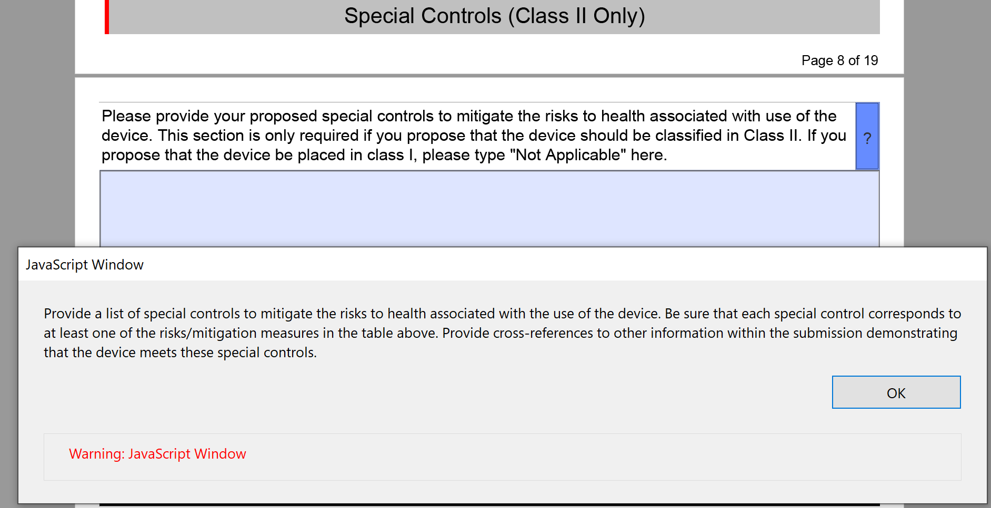
What are your recommended Special Controls?
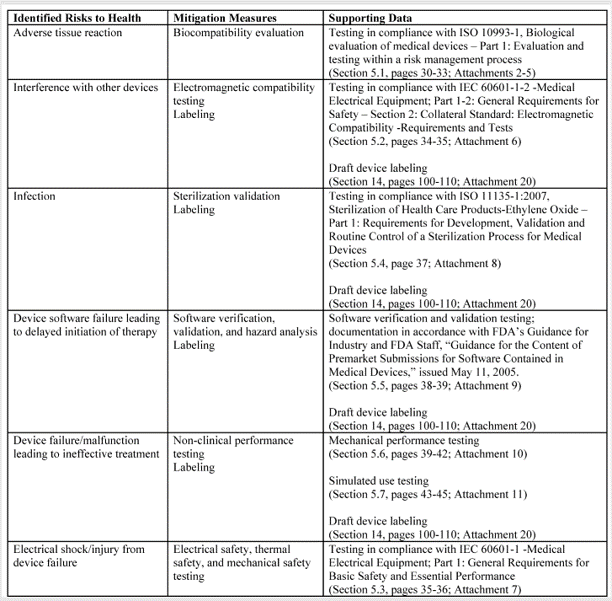
In FDA De Novo Classification Decision Summaries, there is a table provided that identifies the identified risks to health and the recommended mitigation measures for each risk category. In the FDA eSTAR, you are required to add a similar table for De Novo content. The only difference between the table in summary and the eSTAR is that the eSTAR table has a third column where the FDA wants you to reference the supporting data provided for each mitigation measure–including the document and page within the document. The FDA also provided an example table in the eSTAR, copied below.
The above table for the risks to health and mitigations needs to be translated into a list of recommended Special Controls for Class II devices. Since most De Novo applications are for Class II devices, you will need to convert each of your mitigations into a corresponding Special Control and type these controls into the text box provided in the FDA eSTAR.
What else is different from a 510k?
There are no additional mandatory elements that you need to include in a De Novo application, but there are several elements of a 510k submission that are not included in a De Novo. The most obvious of these sections is the Substantial Equivalence Comparison Table in the section labeled “Predicates and Substantial Equivalence.” Another difference is that you are more likely to need clinical data to support a De Novo application than for a 510k submission. It is also possible that subsequent 510k submissions for the same product code may not need to provide clinical data because the 510k process only requires a demonstration of substantial equivalence rather than clinical benefits outweighing risks to health. The FDA review time for a Traditional 510(k) varied between 190 and 210 days in 2022, while the De Novo review timeline averaged 390 days in 2022. Finally, the FDA user fees for 510k submissions are far less than those for a De Novo application.
What in case the product code already exists but you don’t think you will get SE with the existing predicate device? Can you then still go for de novo?
You can always apply for a De Novo, but the first thing the FDA does is to make sure that there is not an existing product code that could be used. If there is, they typically deny the request for De Novo. Therefore, if your product is in the “grey area,” it might make sense to submit a 513(g) to verify in writing that your device requires a De Novo before submitting a De Novo. The 513(g) cost is $3,264 for small businesses, and this gives the FDA resources and information to verify if there is an existing code you should be using instead of requesting a De Novo.
Most people submit just the minimum information in a 513(g). However, we recommend submitting the following additional information to make sure that the FDA has everything they need to determine if your device requires a 510(k) or a De Novo:
1. device description
2. draft instructions for use
3. draft labeling
4. classification rationale as Class 1 or 2
5. a proposed regulation name and number
6. proposed special controls
7. draft benefit/risk analysis
8. risk mitigation table
9. alternative procedures and techniques to the subject device’s technology
10. your efforts to identify a potential predicate device
Rob, I have always wondered what is the difference, in terms of outcomes, between submitting a 513g with this information or just asking the question as part of a pre-sub. Probably, you will not ask “what is the right submission for my product?”, but if instead you ask “based on XYZ, we believe that this product should be submitted through a DeNovo application, does the agency agree with this approach?”, wouldn’t the answer to this question in a pre sub have the same result as a 513g?
The wording I use in a pre-sub for this question is: “Does the FDA agree that the correct regulatory pathway is a 510(k) submission using Kxxxxxx as a potential predicate device?” The FDA is trying to reduce the number of redundant or unnecessary submissions, and therefore they may try to answer your question directly in the pre-sub. If they disagree, the FDA might explain what differences in indications for use or technological characteristics preclude the use of the 510(k) pathway or why your chosen predicate is unsuitable. The answer might include the following conclusion: “For these reasons, and since we are not aware of a previously cleared or approved device with the same indications for use, we believe that a direct De Novo appears to be the most appropriate regulatory pathway for the proposed device. For more information regarding the process for review of De Novo classification requests and documentation required for review, refer to the FDA guidance, “De Novo Classification Process (Evaluation of Automatic Class III Designation).”
The problem with the pre-submission is that the FDA cannot review any data and only has limited time and resources to review your pre-sub documents. With a 513(g) submission the FDA has time and resources to review your documentation more thoroughly and do additional research. Therefore, in borderline cases where the answer is less obvious, a 513(g) may be necessary to get the right answer from the FDA. If the FDA suggests that you consider submitting a 513(g) in their pre-sub response, you should definitely submit a 513(g) before submitting a De Novo.
There is a 3rd approach…you can submit a 510(k) instead of a 513(g). If you get an NSE letter, then you have no choice but to submit a De Novo.