Medical Device Shortage Reporting
The FDA and Health Canada both have executive-level orders requiring medical device shortage reporting or supply-chain disruptions.
In a previous article, we discussed supply-chain disruptions and mentioned that there might be medical device shortage reporting requirements if that disruption causes a market shortage of the manufactured device. Both the United States and Canada have reporting requirements for supply disruptions or the market’s ability to meet the demand of specific types of devices.

Both the U.S. FDA and Health Canada have executive-level orders that require reporting of shortages or disruptions to the supply of medical devices deemed necessary for the COVID-19 Health Emergency. There is some overlap, but each country is monitoring and experiencing shortages and disruptions of different devices.
Where did medical device shortage reporting responsibilities come from?
Check 21 CFR 820, ISO 13485:2016, and even peek at SOR 98-282 and see if you can find your obligations for reporting. Go ahead. I’ll wait… Not much in there, right? Adverse events, complaints, etc., but not market shortages.
Medical device shortage reporting is specific to health emergencies. The U.S. FDA and Health Canada happen to be two authorities having jurisdiction with reporting requirements for shortages concerning the COVID-19 Health Emergency. However, there may be others, so having your organization’s regulatory affairs manager verify the reporting requirements for the markets in which you are engaged might not be bad.
U.S. FDA 506J reporting-
In the United States, an Amendment to the U.S. Food, Drug, and Cosmetics Act requires regulatory reporting by medical device manufacturers to the U.S. FDA. It is sometimes called 506J reporting for the Section of the U.S. FD&C Act where it is located.
You will find the statutory requirements outlined within 21 USC 356J.

For the full text read, 21 USC 356j: Discontinuance or interruption in the production of medical devices. (Interestingly enough, the website where this information is available is not an HTTPS site, so visit at your own discretion).
http://uscode.house.gov/browse.xhtml
What devices are subject to 506J reporting?
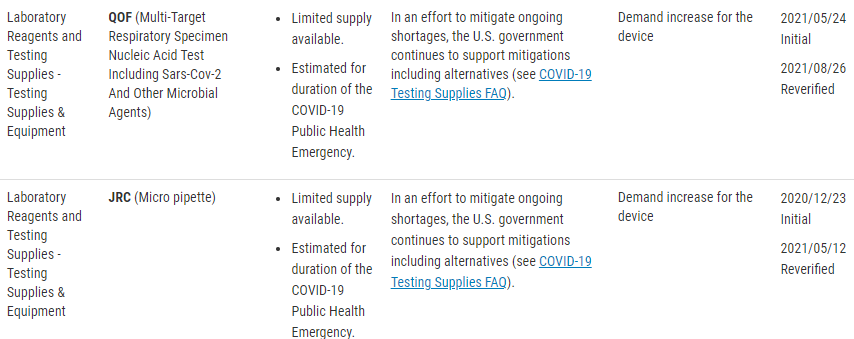
There are two types of devices that the FDA is monitoring. “Critical” devices and an FDA-published list of devices for which COVID-19 is causing a higher than expected demand.
The FDA has released a guidance document that contains criteria for what is considered to be a “Critical Device”. This includes devices such as those used during surgery, emergency medical care, and those intended to treat, diagnose, prevent, or mitigate COVID-19.

There is also a published list of concerned devices that the FDA is specifically monitoring. The FDA website lists these devices by product code, but include the following device types;
- Clinical Chemistry Products
- Dialysis-Related Products
- General ICU/Hospital Products
- Hematology Products
- Infusion Pumps and Related Accessories
- Microbiology Products
- Needles and Syringes
- Personal Protective Equipment (PPE)
- Sterilization Products
- Testing Supplies and Equipment
- Ventilation-Related Products
- Vital Sign Monitoring
Understandably this process may not be intuitive, and for this, the FDA has released a guidance document that addresses;
- Who must make the notification
- When you should make a notification
- What information needs to be included within your 506J notification
- How to make a notification, and
- Penalties for failure to make a notification
The referenced product codes may not be an all-inclusive list or entirely up to date. The best suggestion for full compliance is to go straight to the source of the regulation, in part because noncompliance can result in enforcement action from the FDA. If you think that your device might require notification to the FDA but isn’t in the reference table, you should contact the FDA for notification clarification. Below is the quote from the FDA website, and it includes the contact email for asking these specific questions to ‘the agency.’
“If a device type is not included in this table, but you believe it requires a notification under section 506J of the FD&C Act, or if you have questions regarding the device types in this table, you should contact FDA at CDRHManufacturerShortage@fda.hhs.gov and include “Question” in the subject line of the email.”
Link to the FDA Guidance Document for 506J Reporting- HERE
How to make a 506J report to the U.S. FDA?
The FDA accepts 506J reports in multiple ways. For example, you may use the 506J Reporting web form or submit a notification by email directly to (Include Email Here). In addition, Medical Device Academy has developed a Work Instruction and Form to determine if your company is experiencing a reportable discontinuance or meaningful disruption in manufacturing a medical device as well as compiling the report for submission.
There are a few methods of notification, a web form for individual notifications and spreadsheet options for multiple notifications at once, or emailing a report directly to the FDA reporting email included below;
CDRHManufacturerShortage@fda.hhs.gov

It is for this process that Medical Device Academy developed WI-010 506J Shortage Reporting to the U.S. FDA. This work instruction and associated form, FRM-053 506J Reporting Form are designed to walk you through the process of determining reportability and compiling the information necessary to either complete the webform or email the report directly to the shortage reporting email.
Medical Device Shortage Reporting to Health Canada
Rather than discontinuance and disruption of manufacture, Health Canada is monitoring for shortages of specific devices. Therefore, Health Canada wants Medical Device Shortage Reports regardless of the reason for the shortage. It also shows that this is not identical reporting of the same conditions to two different authorities. Health Canada will also accept reports from Importers because the frame of reference is Canada’s supply of medical devices concerning Canada’s needs.
As an Authority Having Jurisdiction, Health Canada also has reporting requirements for medical device shortage reporting of specific types of medical devices. Health Canada is also an independent authority that uses a different device classification system than the U.S. FDA.
The table below shows the device types by their classification level that HC requires supply chain disruption notifications for. This information is current as of September 5th, 2021, and the link below will take you to the HC website page for the most up-to-date list.
| Class I Medical Devices |
| Masks (surgical, procedure or medical masks) – Level 1, 2, 3 (ATSM) |
| N95 respirators for medical use |
| KN95 respirators for medical use |
| Face shields |
| Gowns (isolation or surgical gowns) – Level 2, 3 and 4 |
| Gowns (chemotherapy gowns) |
| Class II Medical Devices |
| Ventilators (including bi-level positive airway pressure or BiPAP machines, and continuous positive airway pressure or CPAP machines) |
| Infrared thermometers |
| Digital thermometers |
| Oxygen Concentrators |
| Pulse Oximeters (single measurement) |
| Aspirators/suction pumps (portable and stationary) |
| Laryngoscopes |
| Endotracheal tubes |
| Manual resuscitation bags (individually or part of a kit) |
| Medical Gloves – Examination and Surgical (Nitrile, Vinyl) |
| Oxygen Delivery Devices |
| Class III Medical Devices |
| Ventilators (including bi-level positive airway pressure or BiPAP machines) |
| Pulse Oximeters (continuous monitoring) |
| Vital Signs Monitors |
| Dialyzers |
| Infusion Pumps |
| Anesthesia Delivery Devices |
| Class IV Medical Devices |
| Extracorporeal Membrane Oxygenation (ECMO) Devices |
One of the things that Health Canada does an excellent job of is defining its expectations. In the Second Interim Order Respecting Drugs, Medical Devices and Foods for a Special Dietary Purpose in Relation to COVID-19, it is explained the Manufacturers or Importers should report to the Minister actual or expected shortages of the device, OR components, accessories, or parts. These notifications must be made within 5-days of becoming aware of the shortage or the anticipated shortage date. Update reports must be made within 2-days of becoming aware of new information regarding the shortage, and a closing report must be made within 2-days of the end of the shortage.
(This link is to the HC website for the 2nd Interim Order referenced above)
How to make a shortage report to Health Canada?
These reports are submitted online through the Health Canada Website. They have an entire section dedicated to medical device shortages, and the reporting links can be found there (Link here). If you have any questions or are on the fence about notification, you can email Health Canada at MD.shortages-penurie.de.IM@canada.ca.
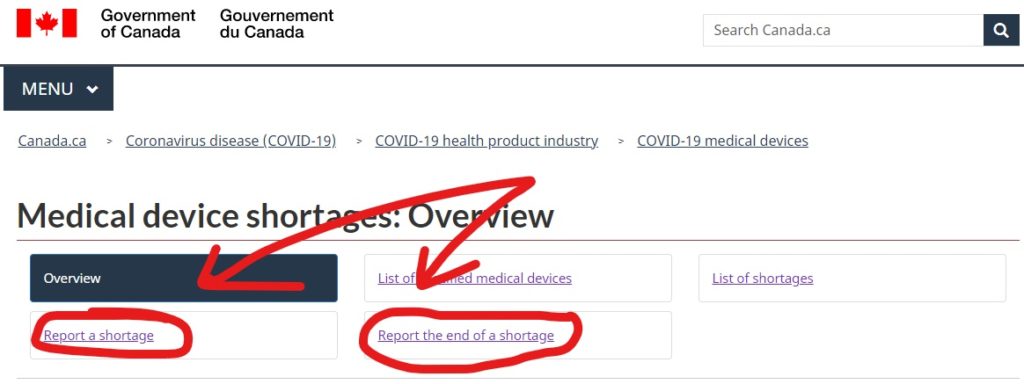
The webform for reporting a shortage is the same webform that is used for providing update reports to Health Canada as well. This is both for manufacturers of specified medical devices as well as importers.
Medical Device Shortage Reporting Read More »





