Your CAPA procedure is the most important SOP. It forces you to investigate quality problems and take actions to prevent nonconformity.
Make sure your CAPA procedure is clear and concise
Procedures are often unclear because the author is more familiar with the process than the intended audience for the procedure. An author may abbreviate a step or skip it altogether. As an author, you should use an outline format and match your CAPA form exactly. There should be nothing extra in the procedure, and nothing left out. Medical Device Academy’s updated CAPA procedure is only six pages and the CAPA form is four pages.

When is the CAPA Procedure Webinar?
The CAPA procedure webinar will be hosted on July 22, 2024 @ 10:30 a.m. ET. The webinar will be live, and you will receive login instructions if you purchased the CAPA procedure prior to the scheduled live webinar. The webinar will also be recorded. You will be able to download the webinar from our Dropbox folder and watch the webinar as many times as needed–including the training of future employees.
What is included with the CAPA Procedure?
The following items are included with the purchase of the CAPA procedure:
- SYS-024, CAPA Procedure
- LST-005, CAPA Log
- FRM-009, CAPA Report
- Native Webinar Slide Deck
- Webinar Recording
This CAPA procedure includes a webinar, but the focus is on implementing the procedure and customizing it for your company. In contrast, the risk-based CAPA training webinar explains the details of performing a CAPA investigation, identifying the root cause, writing a corrective action plan, and performing effectiveness checks. The risk-based CAPA training is recommended for anyone without prior CAPA experience.
CAPA procedure writing recommendations
Procedures are often unclear because the author is an expert with more experience than the intended audience. An author may abbreviate a step or skip it altogether. As an author, you should use an outline format and match your CAPA form exactly. There should be nothing extra in the procedure, and nothing left out. Before writing your own CAPA procedure, consider following these 7 steps:
- Design your CAPA form first
- Identify which steps in your process are most important and specify how these steps will be monitored (i.e., risk-based approach)
- Write a procedure that follows your CAPA form and includes instructions for monitoring and measuring your CAPA process
- Conduct group CAPA training using the draft version of your form and procedure
- Make revisions to the form and procedure to clarify steps the trainees had difficulty with
- Ask the trainees to review the revised form and procedure
- Make final revisions and route the procedure for approval
The specific order of steps is essential to creating a CAPA procedure—or any procedure. Writing a procedure that matches the form used with that procedure helps people understand the tasks within a process. Throughout the rest of this article, we describe each of the nine steps of Medical Device Academy’s CAPA procedure (SYS-024). The actual CAPA form (FRM-009) sold with SYS-024 is more complex than 9 steps, but a more complex form is needed to make sure every sub-task is documented in your CAPA records.
Review nonconformities, including complaints, to determine if a CAPA is needed (Step 1)
If I am auditing a CAPA process, and almost all the CAPAs are resulting from auditor findings, then I know the client is not adequately reviewing other sources of potential quality issues. When I took my first CAPA training course, the image below was drawn on a flip chart by Kim Trautman. I have used this image in all of my CAPA training for other people since. I think this provides a good visual representation of the most common sources of new CAPAs. Although the number of CAPAs from each source will never be equal, you should review all of these data sources for quality issues periodically.
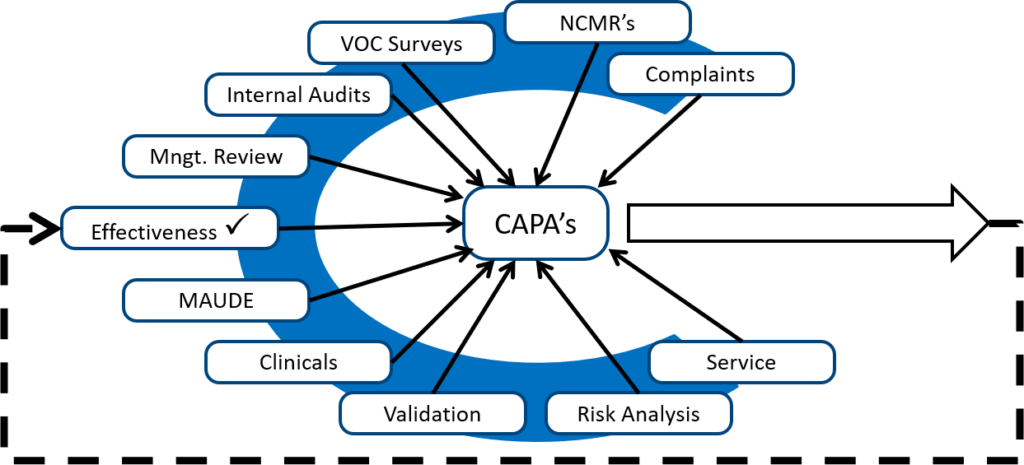
Additionally, there should also be a mix of both ‘Corrective’, and ‘Preventive’ actions. With the FDA’s QSMR transition to harmonize with ISO 13485, this means that they will be looking at both the sub-clause for Corrective Actions, and the sub-clause for Preventive Actions individually.
CAPA procedure step 2 – Describe and reference the quality issue
The next step is to copy and paste your quality issue directly into the CAPA record and add a reference to the source. This step of the CAPA procedure is not a specific requirement of the ISO 13485 standard or the FDA regulations. However, describing and referencing quality issues in your CAPA record is a practical requirement. The person assigned to investigate the root cause of the quality issue needs to know what the source of the quality issue is, and when you are trying to close a complaint or audit report you will find it helpful to cross-reference the two records. For example, the CAPA might be related to the fifth nonconformity in your second internal audit report for 2022 (e.g. IA 220205).

Attribution: Icons were copied and pasted from Flaticon.com.
Step 3 – Perform a root cause analysis
Why can’t we fix our mistakes the first time? We are doomed to repeat mistakes when we fail to identify the root cause or causes. The person you assign to investigate a quality issue must be trained to perform a thorough root cause analysis. Successful root cause analysis depends upon four things:
- Courage to admit that your process is broken
- Learning more than one tool for analyzing problems
- Practicing the use of root cause analysis tools
- Sampling enough records (or testing enough product)
The common belief is that people fail to identify the root cause because they need root cause training (#2) or more practice (#3). However, most people fail because they stop sampling or testing too soon. I typically recommend that companies sample at least twice as many records as the suspected problem frequency. For example, if a complaint occurs 1% of the time, you should review 200 records before you can be sure you identified that root cause. If you are correct, you will only find the quality issue twice in the 200 records. However, a review of 200 records often reveals that the quality problem is more common than you originally estimated and there is more than one cause of device malfunction.
CAPA procedure step 4 – Do you need a new CAPA?
After you have successfully completed a root cause analysis, you now need to determine if a new CAPA is needed. If there is already a CAPA that is open for the same quality issue, you can use the existing CAPA as justification for not conducting corrective actions. In this case, you should include a cross-reference in the new CAPA record to the existing CAPA record. You should also document containment measures and corrections.
In both the existing CAPA record and your new CAPA record you should also be documenting your risk evaluation of your CAPA. In the latest update to our CAPA procedure, we changed the method of risk evaluation to match the MDSAP grading process for nonconformities. A copy of this section is provided in the image below.

In the CAPA procedure, we state that any risk score of 4 or 5 requires the implementation of a CAPA. If any of the escalation rules apply to the risk score, you should implement a CAPA regardless of the total risk score. This is our recommendation for a method of risk evaluation, but there is no standard telling you that you must do it this way. However, we believe this method of calculation is more likely to be consistent because it is based on the MDSAP grading guidelines.
If no escalation rules apply to your risk score, it may be possible to implement containment and corrections only. If your action plan includes only containment and corrections, we recommend that you monitor the quality issue as a process metric or quality objective to identify future occurrences. If you are evaluating a new CAPA that appears to have the same root cause as an existing CAPA, you may need to update the risk score of the existing CAPA to a higher number based on the escalation rules. Escalation may impact your corrective action plan, and it should certainly affect the prioritization of your existing CAPA.
Plan and document your corrective actions, including updating documentation (Step 5)
The biggest mistake you can make in this stage of the CAPA process is to spend too much time planning your corrective actions. “Take action and document it” is the essence of this step in the CAPA procedure. If you spend all of your time planning, then you will never take action. The CAPA plan can and should be edited. Therefore, if you know a procedure needs revision, start revising the procedure immediately. You can always add more corrective actions to your corrective action plan after you write the procedure, but you need to start writing. The second biggest mistake you can make during this stage is failing to document the actions you take. If you don’t document your actions, it’s a rumor, not a record.

Do your planned actions adversely affect regulatory compliance or safety and performance? (Step 6)
One of the required actions for a CAPA is to update your procedure(s) to reflect any process improvements to eliminate the root cause. When you update procedures, you need to make sure procedural changes do not create a regulatory compliance issue. We do this by inserting a cross-reference to each regulatory requirement in our procedures. The cross-reference is then color-coded and we add a symbol for people that are color blind. Symbols also facilitate electronic searches for regulatory requirements.
If corrective actions you implement involve design changes you will need to repeat design verification and design validation to make sure design changes do not impact safety and performance. If corrective actions change your manufacturing or service processes, you will need to repeat process validation to make sure that the process changes do not impact compliance with your design specifications. These recertification and revalidations steps are frequently forgotten, and they represent the biggest challenge for review and approval of design and process changes (described in the video below).
Perform an effectiveness check – Step 7 of your CAPA procedure
Most people verify CAPA effectiveness by verifying that all the actions planned were completed, but this is not a CAPA effectiveness check. An effectiveness check should use quantitative data from your investigation of the root cause as a benchmark. Then you should verify that the performance after corrective actions is implemented resulted in a decrease in the frequency of the quality problem, a decrease in the severity of the quality problem, or both. Ideally, a process re-validation was performed because validation protocols are required to include quantitative acceptance criteria for success.
Step 8 – Record your CAPA results
You are required to record each step of your CAPA procedure in a CAPA record (FRM-009). Therefore, we created a form that is organized in the order of the CAPA process, and then we wrote the CAPA procedure to match the organization of the form. The biggest mistake we see is that the CAPA owner does not update the record to include all of the details until the CAPA plan is completely implemented. This is a mistake. You should be documenting actions when they are taken. When you gather new information, and you need to update your root cause investigation or your corrective action plan, you are allowed to modify the record. You just need to have a system that allows you to keep track of revisions. This is often referred to as an “audit trail.” If you have a paper-based system, you will need to sign and date the document each time you make an addition. If you revise previous entries, you will need to revise and reprint the CAPA record, and then you will need to sign and date the revised and reprinted CAPA record. Ideally, you will have an electronic system with an audit trail, but software budgets are not infinite.
CAPA procedure step 9 – Close the CAPA record
The last step in the CAPA procedure is to close your CAPA record. As with most quality system records, the person responsible for the process should review and approve each record for closure with a signature and date. If the person assigned to the CAPA left sections incomplete or made mistakes in completing the CAPA form, the person that made the mistake should be instructed to correct the mistake, identify that they made a correction, and identify the date of the correction. If a CAPA is not effective, then a cross-reference to the new CAPA that is opened should be documented in the older CAPA record.
CAPA Training
If you are interested in more training on CAPA, you might be interested in purchasing Medical Device Academy’s Risk-Based CAPA webinar. 99% of companies hold off on their training until a procedure is officially released as a controlled document. In my experience, however, these procedures seem to have a lot of revisions made immediately after the initial release. New users ask simple questions that identify sections of procedures that are unclear or were written out of sequence. Therefore, you should always conduct at least one training session with users prior to the final review and approval of a procedure. This will ensure that the final procedure is right the first time, and it will give those users some ownership of the new procedure.
After you train your initial group, and after you make the edits they recommended, ask those trainees to review and edit your changes to the procedure. Sometimes we don’t completely understand what someone is describing, and sometimes maybe only half-listening. Going back to those people to verify that you accurately interpreted their feedback is the most important step for ensuring that users accept your new procedure.
After you approve your new CAPA procedure, make sure everyone in your company is trained on the final version of the procedure. CAPA is a critical process (i.e., “the heart”) in your quality system. Everyone should understand it. You should also provide extra CAPA training for department managers, such as root cause analysis training because they will be responsible for implementing CAPAs assigned to their department. You can use this 7-step process for any procedure, but ensure you use it for the most important process of all—your corrective and preventive action process.
Thank you for reading to the end of this article. Spaz and I thank you for your support.