Redirection of Root Cause Analysis old page
This page has been moved…https://medicaldeviceacademy.com/root-cause-analysis/
Redirection of Root Cause Analysis old page Read More »
The author describes four tools (Five Why Analysis, Is/Is Not Analysis, Fishbone Diagram, and Pareto Analysis) and how each one can help conduct effective root cause analysis.
Quality problems are like weeds. If you don’t pull them out by the root, they grow right back.

Most companies are doomed to repeat their mistakes because the root cause of their mistakes is not fixed. Why don’t companies fix their mistakes? Because the people responsible for the corrective actions (CAPA), were not adequately trained on root cause analysis. Adequate training on root cause analysis requires three things:
If your auditor identifies a nonconformity and you disagree with the finding, then you should not accept the finding and state your case. If an inspector rejects a part, and you believe the part is acceptable, then you should allow the part to be used “as is.” In both of these cases, however, you need to be very careful. Sometimes the problem is that “acceptable” is not as well-defined as we thought. I recommend pausing a moment and reflecting on what your auditors and inspectors are saying and doing. You may realize that you caused the problem.
Once you have accepted that there is a problem, you need to learn how to analyze the problem. There are five root cause analysis tools that I recommend:
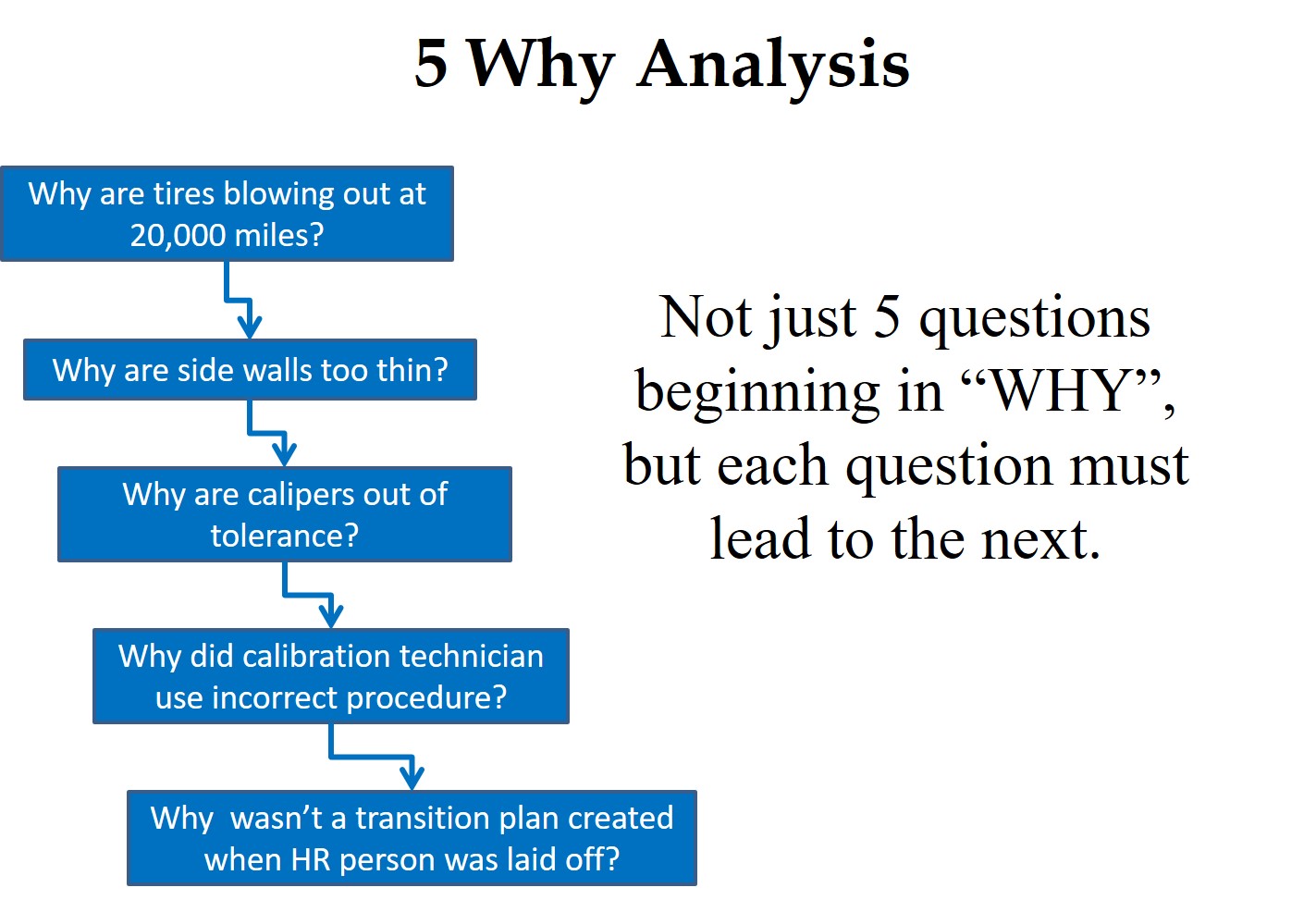
A “Five Why Analysis” is not just five questions that begin with the word “why.” Taiichi Ohno is credited with institutionalizing the “Five Why Analysis” at Toyota as a tool to drill down to the root cause of a problem by asking why five times. I have read about this, used this tool, and taught this concept to students, but I learned of a critical instruction that I was missing when I read Toyota Under Fire.
In that book, Jeff Liker makes the following statement, “Toyota Business Practices dictates using the ‘Five Whys’ to get to the root cause of a problem, not the ‘Five Whos’ to find a fire the guilty party.” At the end of the book, there are lessons learned from Toyota’s experience. Lesson 2 says, “There is no value to the Five Whys if you stop when you find a problem that is outside of your control.” If your company is going to use this tool, it is important that the responsible person is the one performing the five why analysis, and asks why they didn’t take into account forces that are out of their control.
The next tool was presented to me at an AAMI course that I attended on CAPA. One of the instructors was from Pathwise, and he explained the “Pathwise Process” to us for problem-solving. A few years later, I learned that this tool is called the “Is/Is Not Analysis.” This tool is intended to be used when you are having trouble identifying the source of a problem. This method involves asking where the problem is occurring as a potential clue to the reason for the problem. For example, if the problem only occurs on one machine, you can rule out a lot of possible factors and focus on the few that are machine-specific.
The reverse approach is also used to help identify the cause. You can ask where the problem is not occurring. This approach may also lead you to possible solutions to your problem. For example, if the problem never occurs on the first or second shift, you should focus on the processes and the people that work on the third shift to locate the cause. The “Is/Is Not Analysis” is seldom used alone, but it may be the first step toward locating the cause of a quality problem.

This name comes from the shape of the diagram. Other names for this diagram are the “Cause and Effect” or “Ishikawa” diagram. If a problem is occurring in low frequency and has always existed, this might not be your first tool. However, I typically start with this tool when I am doing an investigation of nonconforming product—especially when rejects suddenly appear.
If you are baffled about the cause of a problem, brainstorming the possible causes in a group sometimes works. However, I like to organize and categorize the ideas from a brainstorming session into the “6Ms” of the Fishbone Diagram.
The fourth root cause analysis tool is the Pareto Analysis named after Antoine Pareto. This tool is also a philosophy that was the subject of a book called The 80/20 Principle: The Secret to Achieving More with Less. The Pareto Analysis is used to organize a large number of nonconformities and prioritize the quality problems based upon the frequency of occurrence. The Pareto Chart presents each challenge in descending order from the highest rate to the lowest frequency. After you perform your Pareto Analysis, you should open a CAPA for the #1 problem, and then open a CAPA for the #2 problem. If you get to #3, consider yourself lucky to have the time and resources for it. We have an example of a Pareto Chart in our article on FDA 483 inspection observations from 2013.
If you are interested in learning more about root cause analysis and practicing these techniques, please register for the Medical Device Academy’s Risk-Based CAPA training.
Root cause analysis – Learn 4 tools Read More »
Quality system auditing is outsourced to consultants providing auditing services to ensure auditor independence. Do you need a quote?
The form below provides us with the basic information we need to prepare an auditing services quote for your company. There are instructions below the form that explain exactly what information we are looking for in each section of the form. The quotation process is not automated. A real person (i.e. Lindsey Walker) will get back to you with a quotation. She is our audit program manager. She creates the audit quote and assigns the auditors based on availability and your auditing needs. Her email is sales@medicaldeviceacademy.com. The quotation will be automatically emailed from Freshbooks once she is finished, and then she will follow up with a manual email–just in case your spam filters prevent delivery of the automated email generated by FreshBooks. If Lindsey is on vacation, or out sick, the proposal will be prepared by Rob Packard. His email is rob@fdaestar.com.
If you are looking for the cheapest auditing services you can find, don’t even bother filling in the form. Our goal is to help you improve your quality system and provide valuable consulting advice to achieve improvements. We specialize in helping start-up companies achieve initial ISO 13485 certification, MDSAP certification, and CE Certification. We will assign an experienced lead auditor with an hourly consulting rate of $275/hour. Typically, we will charge $2,750 plus travel expenses for a one-day supplier audit because we expect to spend 30 minutes on audit preparation, eight hours on-site actively auditing, and 2+ hours generating an audit report. Most quotations are flat-fee quotations so you know exactly how much you will be charged. We also request a 50% deposit for audits.
If you are looking for the cheapest auditing services you can find, don’t even bother filling in the form. Our goal is to help you improve your quality system and provide valuable consulting advice to achieve improvements. We specialize in helping start-up companies achieve initial ISO 13485 certification, MDSAP certification, and CE Certification. We will assign an experienced lead auditor with an hourly consulting rate of $275/hour. Typically, we will charge $2,750 plus travel expenses for a one-day supplier audit because we expect to spend 30 minutes on audit preparation, eight hours on-site actively auditing, and 2+ hours generating an audit report. Most quotations are flat-fee quotations so you know exactly how much you will be charged. We also request a 50% deposit for audits.
I need an Audit quote
 In this text box, we need you to identify the process areas you want us to audit. You can ask us to audit just one process or multiple processes. For example, if you are the Quality Manager and the only qualified lead auditor in your company, you might want us to audit your internal auditing, CAPA, management review, control of documents, and control of records. For a single process audit, we generally recommend remote audits via Zoom in order to eliminate the cost of travel. This is also a great way to test us before you engage our firm for a full-quality system internal audit. This is also known as the “audit scope,” and should not be confused with “audit criteria” discussed below. The scope can also include the location of the audit.
In this text box, we need you to identify the process areas you want us to audit. You can ask us to audit just one process or multiple processes. For example, if you are the Quality Manager and the only qualified lead auditor in your company, you might want us to audit your internal auditing, CAPA, management review, control of documents, and control of records. For a single process audit, we generally recommend remote audits via Zoom in order to eliminate the cost of travel. This is also a great way to test us before you engage our firm for a full-quality system internal audit. This is also known as the “audit scope,” and should not be confused with “audit criteria” discussed below. The scope can also include the location of the audit. If you want us to conduct the audit remotely via Zoom, please enter “Remote” in the text box of the auditing services quote form. You can also specify another teleconferencing software of your choice. In general, we recommend that remote audits be split into 90-minute segments or less where one or two processes are covered during the 90-minute Zoom meeting. We explain this further in one of our blog articles: “Why remote audit duration should never exceed 90 minutes.” If you want us to conduct the audit on-site, please provide the address of the audit location and we will include the estimated travel costs in our proposal.
If you want us to conduct the audit remotely via Zoom, please enter “Remote” in the text box of the auditing services quote form. You can also specify another teleconferencing software of your choice. In general, we recommend that remote audits be split into 90-minute segments or less where one or two processes are covered during the 90-minute Zoom meeting. We explain this further in one of our blog articles: “Why remote audit duration should never exceed 90 minutes.” If you want us to conduct the audit on-site, please provide the address of the audit location and we will include the estimated travel costs in our proposal. Please enter the date or dates that you want us to conduct your audit. You can also specify before a specific deadline (e.g. before June 30th). If you want us to conduct an audit of multiple processes remotely, it would help to know what dates and or times of day you would prefer. You can also enter a phone number and say “call me” next to the phone number. Then Lindsey or one of our assigned auditors will contact you to schedule a date and time for your audit.
Please enter the date or dates that you want us to conduct your audit. You can also specify before a specific deadline (e.g. before June 30th). If you want us to conduct an audit of multiple processes remotely, it would help to know what dates and or times of day you would prefer. You can also enter a phone number and say “call me” next to the phone number. Then Lindsey or one of our assigned auditors will contact you to schedule a date and time for your audit.  Please enter the desired duration of the auditing services you want to be quoted. We typically expect at least 30 minutes of audit preparation to review the audit preparation documents that you provide and to create an audit agenda. In addition, we expect to spend approximately two hours of report writing time for each eight-hour day of auditing. Therefore, a typically one-day supplier audit will require a duration of ten hours, while a three-day on-site internal audit will require a duration of 30 hours.
Please enter the desired duration of the auditing services you want to be quoted. We typically expect at least 30 minutes of audit preparation to review the audit preparation documents that you provide and to create an audit agenda. In addition, we expect to spend approximately two hours of report writing time for each eight-hour day of auditing. Therefore, a typically one-day supplier audit will require a duration of ten hours, while a three-day on-site internal audit will require a duration of 30 hours. It is important to specify the audit criteria for your auditing services quote, because otherwise, we might assign an auditor that does not have training on that criteria. Audit criteria are the standards, regulations, procedures, and contracts that may be used to evaluate your quality system or an individual process. Most of our audit team is qualified to audit against the following criteria:
It is important to specify the audit criteria for your auditing services quote, because otherwise, we might assign an auditor that does not have training on that criteria. Audit criteria are the standards, regulations, procedures, and contracts that may be used to evaluate your quality system or an individual process. Most of our audit team is qualified to audit against the following criteria:Auditing Services Quote Read More »
Below is a countdown clock for our next live-stream YouTube video, as well as the schedule for the new live webinars we are hosting.
Our next YouTube Live-streaming video will be:
Medical device cybersecurity testing – Who do you recommend?
🗓️Thursday, August 6, 2026 @ 11:00 a.m. ET
Starts in...
📌For 2026, we are scheduling our live-streaming on Thursdays at 12:30 pm ET.
Do you have suggestions for future webinars and live-streaming videos? Visit our Suggestion Portal.
Our latest YouTube shorts.

If you want to be notified of new webinars another way, including the paid webinars that we do not post on our YouTube channel, please subscribe to our email webinar notification list using the form below:
In addition to the webinars available for purchase on this page, you can also watch videos that we have posted on our YouTube channel. If you are a channel subscriber, you will receive automatic notifications of new YouTube postings on our channel by clicking on the notification bell. The time remaining until our YouTube live-streaming video is shown below, and please don’t forget to email Rob your questions at rob@13485cert.com.
The following is a list of quality and regulatory training webinars that are available for on-demand purchase from this website. If you subscribe to our email notification list using the form on the right, we will notify you by email of any new webinars when we add them to this page. You can also suggest new topics by submitting your idea to the Suggestion Portal.
Webinars Coming Soon Read More »
If you received an FDA 483 inspection observation, make sure you watch this training video before sending your response (within 15 days).

This is the first step in responding to an FDA 483 inspection observation. You need to identify the response deadline. You have 15 business days, starting from the date you received the FDA Form 483 from the inspector. This does not include weekends or US Federal Holidays. To make sure, you can confirm the date with the inspector by email or during the closing meeting before the inspector leaves. If you miss the deadline, and the inspection outcome is “Official Action Indicated (OAI),” then your company will automatically receive a Warning Letter. You and your company’s top management should take this deadline seriously. Seek immediate regulatory help if you do not have extensive experience writing corrective action plans.
Included are examples, recommendations and tips; for implementing the 7 steps, including:
Other related topics reviewed include:
This webinar recording is only $129 (AND INCLUDES NATIVE SLIDE POWERPOINT PRESENTATION FILES):

When you purchase this webinar, you will receive an email with a link for the training video. At the end of the video, there is a slide that provides you with a link to our quiz to verify training effectiveness. The quiz is available on-demand (24/7), and there is no need to download anything. When you submit your answers to the 10 multiple-choice questions, your quiz will be automatically graded, and you will receive the results. If you answer 7 out of 10 questions correctly, you will receive a training certificate. We also have a 10-question quiz for our Risk-Based CAPA webinar.
VIEW OUR PROCEDURES – CLICK HERE OR IMAGE BELOW:


Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a UConn graduate in Chemical Engineering. Rob was a senior manager at several medical device companies, including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, including implants and drug/device combination products for CE marking applications, Canadian medical device license applications, and 510(k) submissions. The favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Facebook.
FDA 483 – 7 Steps to responding to inspection observations Read More »
This CAPA training webinar explains how to create a risk-based CAPA process, and you will learn how to implement corrective and preventive actions step-by-step.Your cart is empty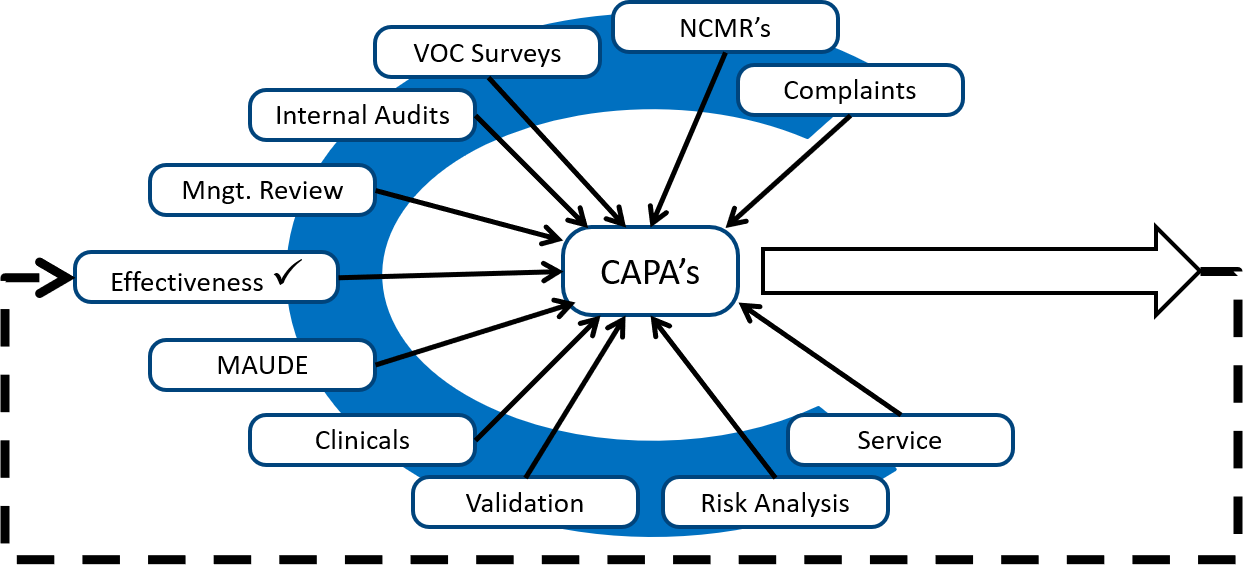
The CAPA training webinar for risk-based CAPA was recorded on June 30, 2020. Everyone who purchases the webinar will receive a link to download the recording and a copy of the native slide deck. We also created a 20-question quiz to verify the training effectiveness. When you purchase the webinar, you will receive links for the slide deck, recording, and the quiz. Upon successful completion of the quiz, you will receive an email that includes your training certificate.
This CAPA training recording is only $129 (AND INCLUDES NATIVE SLIDE POWERPOINT PRESENTATION FILES):
 Create a Risk-Based CAPA ProcessHow to Create a Risk-Based CAPA Process. This webinar includes the most recent updates to ISO 13485:2016, ISO 9001:2015, and ISO 14971:2019.Price: $129.00
Create a Risk-Based CAPA ProcessHow to Create a Risk-Based CAPA Process. This webinar includes the most recent updates to ISO 13485:2016, ISO 9001:2015, and ISO 14971:2019.Price: $129.00
Companies that receive a non-conformity or FDA 483 inspection observation often need a new CAPA procedure as well as CAPA training. Our CAPA procedure, and all of our quality system procedures, are risk-based. Specifically, we use the GHTF grading system to score CAPAs in the same numeric scoring system used for MDSAP audit findings. Therefore, this is the perfect CAPA procedure and form for ISO 13485 certification and MDSAP certification. The CAPA procedure also includes a webinar, but the webinar focus is specific to implementing the procedure and customizing it for your company. In contrast, this risk-based CAPA training webinar explains the details of performing a CAPA investigation, identifying the root cause, writing a corrective action plan, and performing effectiveness checks.
A risk-based CAPA process is a common goal of medical device manufacturers, but until recently “risk-based” was not clearly defined. The two quality management system standards, ISO 9001 and ISO 13485 were revised and re-issued. The current versions are: ISO 9001:2015 released in October 2015 and ISO 13485:2016 released in February 2016. The biggest fundamental change in both standards is an emphasis on risk-based process management. The CAPA process is the heart of your quality system and one of the most important processes. Therefore, this CAPA training gives you a whole new set of tools for managing your CAPA process using a risk-based approach.
This CAPA training goes beyond simple prioritization of CAPAs and color-coding of CAPAs as high, medium, and low risks. Instead, Rob Packard reviews best practices in risk management (i.e., ISO 14971:2019 and ISO/TR 24971:2020) and he applies the more rigorous risk management process to the CAPA process.
This is a “must-see” presentation for anyone that is responsible for the CAPA process or participates in their company’s CAPA Board. Register for our CAPA training and our speaker will help you integrate risk management activities with your CAPA process.
VIEW OUR PROCEDURES – CLICK HERE OR IMAGE BELOW:
 Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.
Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.
CAPA Training – Risk-based Read More »
Can internal auditing be used as a tool specifically designed for conducting a labeling complaint investigation?
A complaint investigation is required when a customer “alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution (21 CFR 820.3b).” In the case of a “labeling complaint,” the customer is alleging that there is a deficiency with the label content, the physical label material itself, or both. In the fictional Halloween story I read about the “Label Monster,” there were five different deficiencies identified in a single unit. The customer refused to accept the entire lot and returned it to the contract manufacturer. “Natalie,” the internal auditor and consultant, was responsible for helping the quality manager (i.e., “Lorelei”) and the operations manager (i.e., “Valerie”) identify the root cause for each of the five label deficiencies. The investigation process involved: 1) interviewing personnel; 2) observing the process, equipment, and environment; and 3) inspecting the returned product.
Routine internal audits are scheduled as part of your annual internal audit schedule. You are the audit program manager, and usually you review your draft annual internal audit schedule with the other members of top management during management review meetings. Since a labeling complaint is not something you can plan or schedule, you would have to amend your internal audit schedule to add a special, “for cause” internal audit of the labeling process to perform a labeling complaint investigation as part of an internal audit. Internal audit scheduling also involves assignment of the lead auditor and team members (if applicable). In the case of labeling at the Red Rum Company, the quality manager was also the audit program manager and responsible for review and approval of the label change control process, inspection of labels prior to use, and final release of each lot. Therefore, an independent lead auditor from outside the company is needed (e.g., a Medical Device Academy auditor).
In addition to differences in the planning of the internal audit, a labeling complaint investigation also reduces the effectiveness of certain parts of the process approach to auditing. Specifically, “Natalie” said, “Reviewing procedures seems like a waste of time.” She was right. Whether the labeling procedure is adequate or not, it is important to evaluate how the process was performed–regardless of what the procedure instructs personnel to do or how well people are trained. Changing procedures and improving training could be a potential corrective action, but correcting procedures and training will not correct the deficiencies observed. We also assumed that the work order content matched the customer purchase order requirements–otherwise the label content would have matched the work order content (i.e., lot numbers should match). Therefore, internal audits performed as part of a complaint investigation should focus on the following elements of the process approach to auditing:
Why didn’t a pFMEA and process validation prevent the labeling errors in our story from happening?
In response to our live-streaming video that was published on Halloween, we received a comment with two questions (copied below).

The story I read was fictional, but in the scenario I was imagining, I envisioned a manufacturing facility that was running extremely well on the two first shift production lines, but nonconformities occurred when production started on second shift for a new product. I envisioned a team consisting of the quality manager (i.e., “Lorelei), operations manager (i.e., “Valerie”), and a possibly a manufacturing engineer and/or quality engineer working together to proactively review the new production line for any potential problems. They started with their pFMEA for the slower first-shift production lines, and modified that pFMEA for the new production line. Unfortunately, the cycle time of the second-shift production line outpaced the capacity of a single labeling machine. Therefore, the team would have three options for addressing this challenge:
All three of the above options are viable, but I imagine most teams would choose the third option. Unfortunately, if your process risk analysis for labeling process controls is designed for an on-demand labeling process, you could easily underestimate the potential complexities of batch building. Even if you evaluate the proposed process controls by conducting a pilot run, production would need to exceed the cycle time of the printers to expose the potential hazardous situation.
The second question that was asked in the YouTube comment would not be relevant, because most of the labeling change control process and the labeling process itself are manual (i.e., no process validation is required, because product is 100% verified). You would still validate that the printer correctly prints the label template in your labeling software, but process validation would not be performed for the manual creation of a label template, manual review of the new label, or manual printing and application of batch-printed labels. Usually, labeling process controls for batch printing of labels focus on initial verification of the label content, verifying the number of labels printed, and reconciling the number of labels printed with the number of labels used. Verification of label content was performed, but only for one label printer–not two printers. There were no problems associated with the number of labels printed or the reconciliation of labels printed with the number used.
In last week’s live-streaming video, we mentioned that an IS/IS NOT analysis would be used as one of the techniques for the labeling complaint investigation. This refers to the process of comparing the areas where a problem is found and where a problem is not found. Problems were not found on product made during the first-shift production, but problems were found on product made during the second-shift production. Additionally, finding problems with half of the product labeling is also critical to identifying the root cause of a failure. Therefore, this tool is extremely valuable when performing a complaint investigation.
A Fish Bone Diagram is another popular root cause analysis tool, because it is a six-part systematic method for identifying a root cause when you don’t know what the cause is. Other names for this tool include: Ishikawa Diagram and Cause and Effect Diagram. The six parts of the method are sometimes called the 6M’s, because you can select words that begin in the letter “M”:
The story read in our live-streaming video was fictional. You rarely have returned product to investigate, you rarely have a supportive team that helps you identify a root cause, and the process rarely as obvious as the story implies. The key to success is hard work and a rigorous investigation that might take more than a week. Rob Packard should not have agreed to a penalty if the CAPA plan was written by the end of the day.
Labeling complaint investigation tool – Internal auditing Read More »
This blog describes best practices for communicating audit findings during an audit, in the closing meeting, and in the audit report.
Would you like to be surprised by an auditor with a major nonconformity? Of course not! Nobody likes that kind of surprise. However, do you know how to effectively communicate your audit findings during the audit, in the closing meeting, and in your audit report?
Audit findings should be communicated at the time the objective evidence is gathered, and it should be clearly stated whether you think the finding is a nonconformity or an opportunity for improvement. Give the auditee an opportunity to correct you.
If you are auditing the process for creating a medical device file, and you are unable to find evidence of product specifications (i.e., ISO 13485:2016, Clause 4.2.3b), then you should restate the requirement and explain why this is a nonconformity. It may be a nonconformity because that requirement is not included in the procedure or index for your medical device file. It may be a nonconformity because the product specification is obsolete and needs to be updated. It may be a nonconformity because you were unable to find the product specification anywhere in the device master record (DMR) index or technical file index. You might also be surprised to learn that product specifications are included in the product user manual, but the process owner forgot that because they were very nervous. The morning after the audit, the process owner may be prepared to show you exactly what you were looking for, including procedural requirements and training.
Some auditors are irritated when they spend time following the audit trail, and after they have taken the time to write a nonconformity, the auditee finally produces the evidence requested. Some auditors say, “It’s too late. You were unable to provide the record when it was requested.” That’s not a value-added finding. The right approach is to say, “Excellent! Now we don’t need to issue a nonconformity or investigate the root cause for a missing product specification.” You might also add, “As a follow-up to this audit, consider ways you can make the product specifications and other required technical documentation easier to find during an audit.” If a similar scenario is repeated during the audit, you might consider writing an OFI beginning with the word “Consider.” However, be careful of suggesting solutions. Medical Device Academy adds cross-references to requirements in each procedure, but that is time-consuming and not required.
In our example above, if evidence of the product specification was not found, that would be a nonconformity. If several other requirements in the medical device file were not available, it would still be a nonconformity. Some people would grade a single lapse as a “minor,” but if multiple requirements are missing they would grade the finding as a “major.” This is not enough to deserve the grading of a “major” but grading subjectivity is difficult to avoid. The specification might exist, but it was accidentally omitted from the file. The specification might not be documented for the file sampled, but it may be easily identified for other product files. The specification might only be missing, because a new employee forgot it and the file was not thoroughly reviewed yet. Therefore, the auditor should consider the missing element an “audit trail.” They should review previous audit reports for similar nonconformities, sample additional requirements, sample other files, and review training records before determining the grading.
In 2012, the Global Harmonization Task Force (GHTF) published a guidance document for grading auditing findings. That guidance proposed a quantitative scoring system with a range of 1-5. Initially, I thought this system was overly complicated. Later, the Medical Device Single Audit Program (MDSAP) adopted the same quantitative scoring system. Since many of our clients adopted MDSAP, we had to learn the MDSAP audit approach and we had to learn how to grade audit findings quantitatively. After using the new system, I realized that the quantitative approach was faster because the objective grading reduced the time required to make a decision on the grade of the finding.
Experienced auditors have most of ISO 13485 memorized, and they usually know which requirements are included in Clauses 4.1-6.3, and which requirements are found later in the standard. Therefore, identifying whether the finding is “direct” or “indirect” is easy. Clauses 4.1-6.3 are indirect clauses, with the exception of 4.2.3 which is direct. There is also one exception to the direct clauses; Clause 8.2.4 is the only clause within Clauses 6.4-8.5.3 that is indirect. It would be easy to persuade someone that there should be additional exceptions, but it would just make the process slower and subjective. Using the clause number for each requirement to determine the initial scoring makes the process faster and more reliable.
There are three escalation rules to consider when grading a nonconformity in the GHTF or MDSAP audit approach. The image below is included in our CAPA form to help remind people of the scoring. The first rule is specific to a repeat nonconformity in the past three (3) years. The second escalation rule is controversial because many people believe the absence of a procedure or records should be sufficient by itself to escalate a finding. However, it’s just a grade, and if the finding is escalated, we want there to be no doubt that the process is not able to meet the requirements. The final escalation rule is the most serious because shipping nonconforming products requires implementation of a recall or field service corrective action (FSCA). Medical Device Academy applies these same three escalation rules when deciding whether a finding is a “major” if a client does not use the MDSAP audit scoring system. This ensures that our grading is objective and it is based on international guidance. We use this same scoring system for internal auditing, supplier auditing, and CAPAs.

In the paragraphs above, we discussed the grading of nonconformities; however, reporting audit findings involves more than just grading nonconformities. ISO 19011:2011 is the official guidance document for auditors of Quality Management Systems, and ISO 13485 is the quality system standard for medical device manufacturers. Section 6.4.2 of this Standard explains best practices for an opening meeting.
The opening meeting is the ideal opportunity to outline how you and your team will present audit findings and to clarify that you will discuss both the strengths and weaknesses of the quality system verbally in the closing meeting and in the audit report. If the auditee is new to auditing, you might even explain the three-part structure of how nonconformities are written.
The option to terminate an audit is typically reserved for a certification audit where multiple major nonconformities are identified, and there is no point in continuing. Termination is highly discouraged because it is better to be aware of all minor and major nonconformities immediately, rather than waiting until the certification audit is rescheduled. The certification body will charge you for their time anyway.
Another reason for termination is when an auditor acts unreasonably or inappropriately. This is rare, but it happens. If the audit is terminated, you should communicate this to upper management at both the certification body and the company, regardless of which side of the table you sit on. For FDA inspections, this is not an option. For audits performed by Notified Bodies, there is the possibility of suspension of a certificate in response to audit termination. Therefore, I always recommend appealing after the fact, instead of termination. Appealing also works for FDA inspections.
The closing meeting should be conducted as scheduled, and the time/location should be communicated to upper management in the audit agenda and during the opening meeting. Top management won’t be happy about nonconformities, but failure to communicate when the closing meeting will be conducted will irritate them further. You should also ensure that a teleconference invitation is set up in advance for the closing meeting, allowing top management to participate remotely if necessary.
At the closing meeting, the auditee should never be taken by surprise. If an issue remains unfulfilled at the closing meeting, the auditee should expect a minor nonconformity—unless the issue warrants a major nonconformity. Since a minor nonconformity can result from a single lapse in fulfilling a requirement, it is challenging for an auditee to argue that an issue does not warrant a minor nonconformity. Typically, the argument is that you are not consistent with other auditors. The most common response to that issue is, “Audits are just a sample, and previous auditors may not have seen the same objective evidence.” The more likely scenario, however, is that the previous auditor interprets the requirements, rather than reviewing them with the client and ensuring both parties agree before a finding is issued.
If a finding is major, the auditee should have very few questions. Additionally, I often find that the reason for a major nonconformity is a lack of management commitment to address the root cause of the problem. Issuing a major nonconformity is sometimes necessary to get management’s attention.
Regardless of the grading, all audit findings will require a corrective action plan—even an FDA warning letter requires a CAPA plan. Therefore, a major nonconformity is not a disaster. You just need to create a more urgent plan for action.
All guides and auditees should be informed of potential findings at the time an issue is identified. This is important so that an auditee has the opportunity to clarify the evidence being presented. Often, nonconformities result from miscommunication between the auditor and the auditee. This often occurs when the auditor lacks a thorough understanding of the process being audited. It is a tremendous waste of time for both sides when this occurs. If there is an actual nonconformity, it is also important to gather as much objective evidence as possible for the auditor to write a thorough finding and for the auditee to prepare an appropriate corrective action plan in response to the discovery.
As an auditor, I encourage auditees to provide honest feedback directly to me and to management, so that I can continue to improve. If you are providing feedback about an internal auditor or a supplier auditor, you should always give feedback directly to the person before going to their superior. You are both likely to work together in the future, and you should give the person every opportunity to hear the feedback firsthand.
When providing feedback from a third-party certification audit, you should know that there will be no negative repercussions against your company if you complain directly to the certification body. At most, the certification body will assign a new auditor for future audits and investigate the need for taking action against the auditor. In all likelihood, any action taken will be “retraining.” I never fired somebody for a single incident—unless they broke the law or did something unsafe. The key to providing feedback, however, is to be objective. Give specific examples in your complaint, and avoid personal feelings and opinions.
As an auditor, one of the most important (and difficult) things to learn is how to issue a nonconformity—especially a major. This is typically done at the closing meeting of an audit; however, the closing meeting is not where the process of issuing the nonconformity begins. Issuing a nonconformity starts in the opening meeting.
As the auditee, you should ask for the contact information of the certification body during the opening meeting. Ask with a smile—just in case you disagree, and so you can provide feedback (which might be positive). As the auditor, you should always provide the certification body’s contact information (if they are a third-party auditor). If you are conducting a supplier audit or an internal audit, you probably know the auditor’s boss, and there is perhaps no formal complaint or appeals process. In the case of a supplier audit, the customer is always right—even when they are wrong.
If you would like to learn more about auditing methods and best practices, consider registering for our Lead Auditor Training Course.
Audit Findings – How to communicate good and bad findings. Read More »
Learn how to become ISO 13485 certified while avoiding the stress that tortures other quality system managers.
ISO 13485 is an international standard for quality management systems that is specific to the medical device industry. ISO 13485:2016 is the most recent version of the standard, and it has become the blueprint for medical device company quality systems globally. If your company wants to design, manufacture, or distribute medical devices, you should consider becoming ISO 13485 certified.
Yes, you need to maintain a copy of the ISO 13485 standard as a “document of external origin.” This is needed for reference when you are making updates to procedures in your quality system. If you are looking for the best place to purchase a copy of the ISO 13485:2016 standard, we recommend the Estonian Centre for Standardisation and Accreditation. If you purchase a copy, we recommend selecting the option for a multi-user license so the standard can be used by more than one person in your company and printed. The only difference between the EN ISO version and the International ISO version is that the EN ISO version includes harmonization Annex ZA for compliance with the EU MDR and Annex ZB for compliance with the EU IVDR. This version is also referred to as A11:2021. Here’s a copy of the text from the beginning of the Standard:
“This Estonian standard EVS-EN ISO 13485:2016/A11:2021 consists of the English text of the European standard EN ISO 13485:2016/A11:2021. This standard has been endorsed with a notification published in the official bulletin of the Estonian Centre for Standardisation and Accreditation. Date of Availability of the European standard is 08.09.2021. The standard is available from the Estonian Centre for Standardisation and Accreditation.”

Rob Packard created his first quality system in the Spring of 2004. In October 2009, after successfully managing quality systems for three different medical device manufacturers, Rob joined BSI as a Lead Auditor and instructor. In April 2010, he purchased the 13485cert.com URL and he began to help companies implement quality systems as a consultant (while continuing to audit and train 140 days per year for BSI). In 2011 his medical device blog postings began as a way to help medical device companies. In 2012, Rob began building a library of quality system procedures for a turn-key quality system and selling the procedures from the Medical Device Academy website. Dozens and dozens of consulting clients have successfully achieved ISO 13485 certification with Medical Device Academy’s turnkey quality system procedures, and hundreds of quality systems were audited and/or improved. This ISO 13485 training webinar is also included as part of our turnkey quality system.
On February 23, 2022, the FDA published a proposed rule for medical device quality system regulation amendments. The FDA planned to implement amended regulations within 12 months, but the consensus of the device industry is that a transition of several years would be necessary. In the proposed rule, the FDA justifies the need for amended regulations based on the “redundancy of effort to comply with two substantially similar requirements,” creating inefficiencies. The FDA also provided estimates of projected cost savings resulting from the proposed rule. What is completely absent from the proposed rule is any mention of the need for modernization of device regulations.
On January 31, 2024, the FDA issued a final rule amending the device current good manufacturing practice (CGMP) requirements of the Quality System (QS) Regulation under 21 CFR 820 to align more closely with ISO 13485. The FDA will begin enforcing this on February 2nd, 2026.
The QSR is 26 years old, and the regulation does not mention cybersecurity, human factors, or post-market surveillance. Risk is only mentioned once by the regulation, and software is only mentioned seven times. The FDA has “patched” the regulations with guidance documents, but there is a desperate need for new regulations that include critical elements. The FDA has “patched” the regulations through guidance documents, but there is a desperate need for new regulations that include critical elements. The transition of quality system requirements for the USA from 21 CFR 820 to ISO 13485:2016 will force regulators to establish policies for compliance with each of these quality system elements. Companies that do not already have ISO 13485 certification should be proactive by 1) updating their quality system to comply with the standard and 2) adopting the best practices outlined in the following related standards:
This 2-part webinar has been previously recorded three different times. Our previous webinar on the 2003 version of ISO 13485 was split into two parts: Stage 1 and Stage 2. That first webinar was recorded in 2015. The webinars were updated in 2016 and again in 2018. We followed the same format, 2-part Stage 1 and Stage 2, for all of the subsequent ISO 13485 training webinars. Stage 1 is a preparatory review, whereas Stage 2 is the in-depth validation for actual certification. Stage 1 is usually 1 to 1.5 days and can be conducted remotely. Stage 2 is usually 3 or more days in duration.
The Stage 1 certification audit training focuses on the following processes:
The Stage 2 certification audit training focuses on the clauses of the ISO 13485:2016 Standard that were not covered in the Stage 1 readiness audit:
This new version of ISO 13485:2016 training will be delivered as a free live-streaming video. After live-streaming the videos, they will remain available to watch as refresher training. The dates of the training sessions are:
This pair of ISO 13485 training webinars explains precisely what you need to do to implement a quality system compliant with ISO 13485. After you creating your own plan (a free template is provided of you register to take the training quiz associated with this ISO 13485:2016 training. You can watch these two webinars with your team so they can implement your plan in the next several months. All deliveries of content are through our streaming channels (e.g., YouTube).
After completing each training webinar, you can verify the training effectiveness and obtain training records from us if you complete the registration form for this webinar above. You can watch the training anytime, watch as many times as you wish, and you can complete the training quizzes anytime. The quizzes will be available after each live-streaming video airs.

There is a big difference between being ISO 13485 certified and being compliant with ISO 13485:2016, the medical devices quality management systems standard. Anyone can claim compliance with the standard. Certification, however, requires that an accredited certification body has followed the requirements of ISO 17021:2015, and they have verified that your quality system is compliant with the standard. To maintain that certification, you must maintain your quality system’s effectiveness and endure both annual surveillance audits and a re-certification audit once every three years.
Step 1 – Planning for ISO 13485 certification
There are six steps in the ISO 13485 certification process, but that does not mean there are only six tasks. The first step in every quality system is planning. Most people refer to the Deming Cycle or Plan-Do-Check-Act (PDCA) Cycle when they describe how to implement a quality system. However, when you are implementing a full quality system, you need to break the “doing” part of the PDCA cycle into many small tasks rather than one big task. You also can’t implement a quality system alone. Quality systems are not the responsibility of the quality manager alone. Implementing a quality system is the responsibility of everyone in top management.
Below you will find seven tasks listed. I did NOT identify these nine tasks as “Steps” in the ISO 13485 certification process, because these tasks are typically repeated for each process in your quality system. Most quality systems are implemented over time, and the scope of the quality system usually grows. Therefore, you are almost certain to have to perform all of the following nine tasks multiple times–even after you receive the initial ISO 13485 certification. As the saying goes, “How do you eat an elephant? One bite at a time.” Therefore, avoid the inevitable heartburn caused by trying to do too much at one time. Implement your quality system one “bite” at a time.
The first task in implementing an ISO 13485 quality system is to purchase a copy of the ISO 13485:2016 standard, such as the MDSAP Audit Approach document. You will also need other applicable medical device standards. Some of these standards are general standards that apply to most, if not all, medical devices, such as ISO 14971:2019 for risk management. There are also guidance documents that explain how to use these general standards, such as ISO/TR 24971:2020, and guidance on how to apply ISO 14971:2019. Finally, there are testing standards that identify testing methods and acceptance criteria for things such as biocompatibility and electrical safety. You will need to monitor these standards for new and revised versions. When these standards are updated, you will need to identify the revised standard and develop a plan for addressing the changes.
When you purchase a standard, be sure to buy an electronic version of the standard so you can search the standard for keywords efficiently. You should also consider purchasing a multi-user license for the standard because every manager in your company will need to look up information in the standard. Alternatively, you could buy a paper copy of the standard and locate the standard where everyone in your company can access it. Often I am asked what the difference is between the EN version of the standard and the ISO version of the standard. “EN” is an abbreviation meaning European Standards or “European Norms,” which is based upon the literal translation from the French (i.e., “normes”) and German (i.e. “norm”) languages. “ISO” versions are international standards. In general, the body of the standard is typically identical but harmonized EN standards for medical devices include annexes ZA, ZB, and ZC that identify any deviations from the requirements in three medical device directives (i.e., MDD, AIMD, and IVDD).
Clause 1 of ISO 13485 is specific to the scope of a quality system. ISO 9001, the general quality system standard, allows you to “exclude” any clause from your quality system certification. However, ISO 13485 will only allow you to exclude design controls (i.e., clause 7.3). Other clauses within ISO 13485 may be identified as “non-applicable” based on the nature of your medical device or service. You must also document the reason for non-applicability in your quality manual. Typically, the following clauses are common clauses identified for non-applicability:
The third task is to assign a process owner to each of the processes in your quality system. Typically, you create a master list of each of the required processes. Usually, the assignments are made to managers in the company who may delegate some or all of a specific process. You should expect most managers to be responsible for more than one process because there are 28 required procedures in ISO 13485:2016, but most companies have fewer than ten people when they first implement a quality system.
The fourth task is to identify which processes need to be created first and to schedule the implementation of procedures from first to last. You can and should build flexibility into the schedule, but some procedures are needed at the beginning. For example, you need document control, record control, and training processes to manage all of your other procedures. You also need to implement the following processes to document your Design History File (DHF): 1) design controls, 2) risk management, 3) software development (if applicable), and 4) usability. Therefore, these represent the seven procedures that most companies will implement as early as possible. Procedures such as complaint handling, medical device reporting, and advisory notice procedures are usually reserved for last. These procedures are last because they are not needed until you have a medical device in use.
Forms create the structure for records in your quality system, and a well-designed form can reduce the need for lengthy explanations in a procedure or work instruction. Therefore, you should consider developing forms first. The form should include all required information that is specified in the applicable standard or regulations, and the cells for that information should be presented in the order that the requirements are listed in the standard. You might even consider numbering the cells of the form to provide an easy cross-reference to the corresponding section of the procedure. Once you create a form, you might consider creating a flowchart next. Flowcharts provide a visual representation of the process. You might consider including numbers in the flow chart that cross-reference to the form as well.
Once you have created a form and a flowchart, you are now ready to write your quality system procedure. Many sections are typically included in a procedure template. It is recommended that you use a template to ensure that none of the basic elements of a procedure are omitted. You might also consider adding two sections that are uncommon to a procedure: 1) a risk analysis of the procedure with the identification of risk controls to prevent risks associated with the procedure, and 2) a section for monitoring and measurement of the process to objectively measure the effectiveness of the process. These metrics are the best sources of preventive actions, and some of the metrics might be potential quality objectives to be identified by top management.
Most companies rely upon internal audits to catch missing elements in their procedures. However, audits are intended to be a sampling rather than a 100% comprehensive assessment. Therefore, when a draft procedure is being reviewed and approved for the first time, or a major rewrite of a procedure is conducted, a thorough gap analysis should be done before the approval of the draft procedure. Matthew Walker created an article explaining how to conduct a gap analysis of procedures. In addition, Matthew has been gradually adding cross-references to ISO 13485:2016 requirements in each procedure. He is color-coding the cross-referenced clauses in blue font as well. This makes it much easier for auditors to verify that a procedure is compliant with the regulations with minimal effort. The success of these two methods has taught us the importance of conducting a gap analysis of all new procedures.
You are required to document the training requirements for each person or each job in your company. Documentation of training requirements may be in a job description or within a procedure. In addition to defining who should be trained, you also need to identify what type of training should be provided. We recommend recording your training to ensure that new future employees receive the same training to ensure consistency. Design controls training should be the first priority. You are also required to maintain records of the training. You must verify that the training was effective, and you need to check whether the person is competent in performing the tasks. This training may require days or weeks to complete. Therefore, you may want to start training people several weeks before your procedure is approved. Alternatively, you can swap the order of tasks and conduct training after the procedure approval. If that approach is taken, then the procedure should indicate the date the procedure becomes effective–typically 30 days after approval to allow time for training.
Approval of a procedure may be accomplished by signing and dating the procedure itself, while another approach is to create a document that lists all the procedures and forms being approved at one time. The second method is the method we use in our turnkey quality system. Companies can review and approve as many procedures at one time as they wish. Since this process needs to be defined to ensure that all of the procedures you implement are approved, the document control process is typically the first procedure that companies will approve in a new quality system. The second procedure generally is for the control of records. Then the next procedures implemented will typically be focused on the documentation of design controls, risk management, usability testing, and software development. The last procedures to be approved are typically complaint handling, medical device reporting, and recalls. These procedures are left for last because you don’t need them until you are selling your medical device.
The last task required for the implementation of a new quality system is to start using the procedures to generate records. All of the procedures will need records before the process can be verified to be effective. Records can be paper-based, or the records can be electronic. Whichever format you use for the record retention needs to be communicated to everyone in the company through your Control of Records procedure and/or within each procedure. If you include the information in each procedure, the records of each procedure should be listed in the procedure, and the location where those records are stored should be identified. Generally, there is no specific minimum number of records to have for a certification audit, but you should have at least a few records for each process that you implement.
Step 2 – Conducting your first internal audit
The purpose of the internal audit is to verify the effectiveness of the quality system and to identify nonconformities before the certification body auditor finds them. To successfully achieve this secondary objective, it is essential to have a more rigorous internal audit than you expect for the certification audit. Therefore, the internal audit should be of equal duration or longer in duration than the certification audit. The internal audit should not consist of a desktop review of procedures. Reviewing procedures should be part of gap analysis (i.e., task 6 above) that is conducted on draft procedures before they are approved. Internal audits should utilize the process approach to auditing, and the auditor should apply a risk-based approach (i.e., focus on those processes that are most likely to contribute to the nonconforming products, result in a complaint, or cause severe injuries and death).
After your internal audit, you will receive an internal audit report from the auditor. You should also expect findings from the internal auditor, and you should expect opportunities for improvement (OFI) to be identified. Experienced auditors can typically identify the root cause of a nonconformity more quickly than most process owners. Therefore, it is recommended for each process owner and subject matter expert to review nonconformities with the auditor and discuss how the nonconformity should be investigated. The root cause must be correctly identified during the CAPA process, and the effectiveness check must be objective to ensure that problems do not recur.
Corrective actions should be initiated for each internal audit finding immediately, to make sure the findings are corrected and prevented from repeat occurrence before the Stage 1 audit. It will take a minimum of 30 days to implement the most corrective actions. Depending upon the scheduling of the internal audit, there may not be sufficient time to complete the corrective actions. However, you should at least initiate a CAPA for each finding, perform an investigation of the root cause, and begin to implement corrective actions.
Also, to take corrective actions related to internal audit findings, you should look for internal audits from other sources. The diagram below shows several different sources of potential corrective and preventive actions.
Monitoring and measuring each process is the best source of preventive actions, while internal audits are typically the best source of corrective actions. Any quality problems identified during validation are also excellent sources of corrective actions because the validation can be repeated as a method of demonstrating that the corrective actions are effective. However, your ISO 13485 certification auditor will focus on non-conforming products, complaints, and services as the most critical sources of corrective actions. These three sources are prioritized because these three sources have the greatest potential for resulting in serious injury, death, or recall if corrective actions are not implemented to prevent problems from recurring.
In addition to completing a full quality system audit before your stage 1 audit, you are also expected to complete at least one management review. To make sure that you have inputs for each of the 12 requirements in the ISO 13485:2016 standard, it is recommended to conduct your management review only after you have completed your full quality system audit and initiated some corrective actions. If possible, you should also conduct supplier audits for any contract manufacturers or contract sterilizers. It is recommended to use a template for that management review that is organized in the order of the required inputs to ensure that none of the necessary inputs are skipped. Quality objectives will need to be established long before the management review so that the top management team has sufficient time to gather data regarding each of the quality objectives. Also, you should consider delegating the responsibility for creating the various slides for each input to different members of top management. This will ensure that everyone invited to the meeting is engaged in the process, and it will spread the workload for meeting preparation across multiple people.
At the end of the meeting, top management will need to create a list of action items to be completed before the next management review meeting. Meeting minutes will need to be documented for the meeting, including the list of action items and each of the four required outputs of the management review process. We recommend using the notes section of a presentation slide deck to document the meeting minutes related to each slide. Then the slide deck can be converted into notes pages and saved as a PDF. The PDF notes pages will be your final meeting minutes for the management review. An example of one of these note pages is provided in the figure below.

One of the more common non-value-added findings by auditors is when an auditor issues a nonconformity because you do not have your next internal audit and your next management review scheduled–even though each may have occurred only a month prior to the Stage 1 audit. Therefore, we recommend that you document your next 12-month cycle for internal audits and schedule your next management review as action items in every management review meeting. The schedule can be adjusted if needed, but this allows top management to emphasize various areas in internal audits that may need improvement. You might even set a quality objective to conduct a minimum of three management reviews per year at the end of your first management review.
In 2006, the ISO 17021 Standard was introduced for assessing certification bodies. This is the standard that defines how certification bodies shall go about conducting your initial certification audit, annual surveillance of your quality system, and the re-certification of your quality system. In the past, certification bodies would typically conduct a “desktop” audit of your company before the on-site visit to make sure that you have all the required procedures. However, ISO 17021 requires that certification bodies conduct a Stage 1 audit that assesses the readiness of your company before conducting a Stage 2 audit. Therefore, even if the Stage 1 audit is conducted remotely, the certification body is expected to interview process owners and sample records to verify that the quality system has been implemented. Certification body auditors will also typically verify that your company has conducted a full quality system audit and at least one management review. Finally, the auditor will usually select a process such as corrective action and preventive action (CAPA) to make sure that you are identifying problems with the quality system and taking appropriate measures to address those problems.
Your goal for the Stage 1 audit should not be perfection. Instead, your focus is to make sure that there are no “major” non-conformities. The term “major” used to have a specific definition:
Under the MDSAP, the grading system for nonconformities now uses a numbering system for grading nonconformities: “Nonconformity Grading System for Regulatory Purposes and Information Exchange Study Group 3 Final Document GHTF/SG3/N19:2012.” Any nonconformity is graded on a scale of one to four, and then two potential escalation rules are applied. If any nonconformities are graded as a four or a 5, then the auditor must assess whether a five-day notice to Regulatory Authorities is required. A five-day notice is required in either of the following situations: 1) one or more findings grading of “5”; or 2) three or more findings graded as “4.” If your Stage 1 audit results in a five-day notice, then you are not ready for your Stage 2 audit. For example, a complete absence of two required procedures in clauses 6.4 through 8.5 of ISO 13485:2016 would result in two findings with a grading of “4.” This would not result in a five-day notice, but the absence of a third required procedure would result in a five-day notice.
The duration of your Stage 1 audit will be one or two days, but a 1.5-day audit is quite common for MDSAP Stage 1 audits. The reason for the 1.5-day Stage 1 audit is that it is challenging to assess readiness for Stage 2 in one day, and if the total duration of Stage 1 and Stage 2 is 5.5 days, then the Stage 2 audit could be completed in four days. The four-day audit is more convenient than a three-day audit for a two-person audit team.
After your Stage 1 audit, you will receive an audit report, and you should expect findings. You should initiate corrective actions for each finding immediately, to make sure the findings are corrected and prevented from repeat occurrence before the Stage 2 audit. The duration between the audits is typically about 4-6 weeks. That does not leave much time for you to initiate a CAPA, perform an investigation of the root cause, and implement corrective action. At a minimum, you must submit a corrective action plan for each finding to your MDSAP auditing organization (AO) within 15 calendar days of receiving the finding. For any findings graded as a “4” or higher, you will need to provide evidence of implementing the corrective action plan to the AO within 30 calendar days of receiving the finding. You are also unlikely to have enough time to conduct an effectiveness check prior to the Stage 2 audit.
The Stage 2 initial ISO 13485 certification audit will verify that all regulatory requirements have been met for any market you plan to distribute in. The auditor will complete an MDSAP checklist that includes all of the regulatory requirements for each of the countries that recognize MDSAP: 1) the USA, 2) Canada, 3) Brazil, 4) Austria, and 5) Japan. The auditor will also sample records from every process in your quality system to verify that the procedures and processes are fully implemented. This audit will typically be at least four days in duration unless multiple auditors are working in an audit team.
The audit objectives for the Stage 2 ISO 13485 certification audit specifically include evaluating the effectiveness of your quality system in the following areas:
All procedures will be reviewed for compliance with ISO 13485:2016 and the applicable regulations. The auditor will also sample records from each process. If the auditor identifies any nonconformities during the audit, it is important to record the findings and begin planning corrective actions immediately. If you have any questions regarding the expectations for the investigation of the root cause, corrections, corrective actions, and effectiveness checks, you should ask the auditor during the audit or the closing meeting. At a minimum, you must submit a corrective action plan for each finding to your MDSAP auditing organization (AO) within 15 calendar days of receiving the finding. For any findings graded as a “4” or higher, you will need to provide evidence of implementing the corrective action plan to the AO within 30 calendar days of receiving the finding. The auditor will not be able to recommend you for ISO 13485 certification until your corrective action plans are accepted.
If you receive a finding with a grading of “5,” or three or more findings graded as “4,” then the MDSAP auditor is required to issue a five-day notification to the regulators. The auditor will also need to return to your facility for a follow-up audit to close as many findings as they can. It is not necessary to eliminate all of the findings in order to be recommended for ISO 13485 certification, but the grading of the findings must be reduced to at least a “3” before recommending the company for certification. The number of findings also determines whether the auditor recommends your company for certification.
In addition to reviewing the findings and conclusions of the audit during the closing meeting, the auditor will also review the plan for the annual surveillance and re-certification with you. Each certification cycle is three years in duration. There will be two surveillance audits of approximately one-third of the duration of the combined duration of stage 1 and stage 2 initial certification audits, and the first surveillance audit must be completed within 12 months of the initial certification audit. In the third year, there will be a re-certification audit for two-thirds of the duration of the combined duration of stage 1 and stage 2 initial certification audits. The initial ISO 13485 certificate will be issued with a three-year expiration, and the certificate is typically received about one month after the acceptance of your corrective action plan.
There are no stupid questions, and we can save you weeks of wasted time if you just ask for help. We are always looking for new ideas for blogs, webinars, and videos on our YouTube channel. If you have any general questions about obtaining ISO 13485:2016 certification, please email Rob Packard at rob@fdaestar.com. If you have a suggestion for new ISO 13485 training materials, you can also use our “Suggestion Portal.” You can also schedule an initial free consultation with Rob using his calendly link.
Rob Packard is a regulatory consultant with 30+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, or Instagram.
ISO 13485 – Need training? Read More »
90% of usability testing submitted to the FDA is unacceptable and the root cause is simply a failure to understand the human factors process.
If you submitted no usability testing to the FDA in your 510(k) submission, it would be obvious why the FDA reviewer identified usability as a major deficiency. However, you spent tens of thousands of dollars on usability testing that delayed the 510(k) submission by six months. Despite all of the time and money your company invested in the human factors process, it appears that you need to start over and repeat the entire process again. The CEO is furious, and he wants you to show him where in the 49-page FDA guidance it says that you have to do things differently.
Unlike CE Marking technical files, the FDA does not require a usability engineering file for all products. Instead, the FDA determines if usability testing is required based on a comparison of your device’s user interface and a competitor’s user interface (i.e. predicate device user interface). If the user interface is identical, then usability testing may not be required. Instead, your company should be able to write a rationale for not doing usability testing based on equivalence with the predicate device. If there are differences in your user interface, you will need to provide use-related risk analysis (URRA), identify critical tasks, implement risk controls, and provide verification testing to demonstrate the effectiveness of the risk controls. Even if your device is “easier to use” or “simpler”, you still need to provide the documentation to support this claim in your submission. The FDA also does not allow comparative claims in your marketing for 510(k) cleared devices. Comparative claims require the support of clinical data.
There is a YouTube video describing these 10 steps at the bottom of this blog posting.
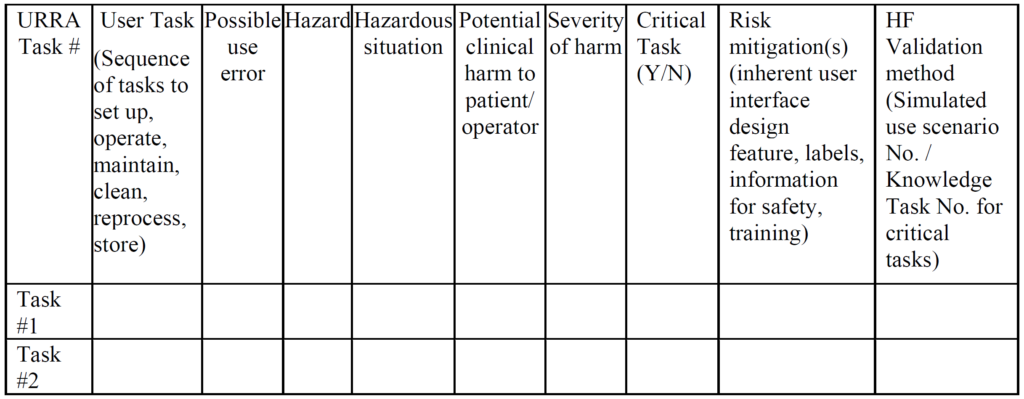
The primary problem with using an FMEA or Fault-Tree risk analysis tool is that these tools involve the estimation of the severity of harm and the probability of occurrence of harm, while the FDA does not feel it is appropriate to estimate the probability of occurrence of harm. Instead, the FDA instructs companies to assume that use errors will occur and to implement risk controls to mitigate those risks (see URRA example above). Although “mitigation” is unlikely, and use risks will only be reduced, this is the approach the FDA wants companies to use. In addition, the FDA expects your company to provide traceability of risk control implementation to each use-related risk you identified and the FDA expects documentation of verification testing (i.e. usability testing) that shows your risk controls are effective. Finally, the FDA (and ISO 14971, Clause 10) expects you to collect and perform a trend analysis of use errors. Any use errors that are reported should be evaluated for the need to implement additional corrective actions to prevent future use errors. Blaming “user error” is not an acceptable approach.
Therefore, if you are developing imaging software, you need to make sure your user group includes radiologists that cover the entire range of competencies. In addition, most radiology images are taken by radiology technicians and then reviewed by the radiologist. Therefore, radiology technicians should be considered a completely different user group due to the differences in experience, training, and competency when compared to a radiologist. This simple example doubles the number of users needed because you have two user groups instead of one.
Human factors process, can we make this easy? Read More »