Learn how to create a UDI procedure for compliance with the FDA and EU regulatory requirements for UDI compliance.
What’s included in our UDI Procedure?
The UDI procedure complies with ISO 13485:2016 as well as the European and US regulations. The procedure includes the following list of documents:
- SYS-039 A D5 UDI Requirements Procedure
- FRM-016 A D1 FDA UDI Checklist
- FRM-017 A D2 EU UDI Checklist
We are including a training webinar explaining the FDA’s UDI System and the native presentation slide deck, and we will provide an exam (i.e., a 10-question quiz) to verify training effectiveness. If you submit the completed exam to us by email in the native MS Word format, we will correct the exam and email you a training certificate with your corrected exam. The FDA website also provides information about the UDI system. We also provide email notifications of free updates to the procedure and forms when we update the procedure to comply with new and revised regulations.


What countries does this UDI procedure apply to?
A Unique Device Identifier or UDI is required for all in vitro diagnostics (IVD) and devices in the USA and Europe to identify the manufacturer, the device or IVD itself, and production-related details such as the date of manufacture and the lot number. To comply with these UDI requirements, you will need a UDI procedure compliant with the US regulations (i.e., 21 CFR 830 and parts of 21 CFR 801). To comply with European regulations, you will need a UDI procedure compliant with Article 24 and Annex VI of the IVDR and Article 27 and Annex VI of the MDR. The video below provides an overview of Medical Device Academy’s UDI procedure.
What is a UDI?
UDI stands for ‘Unique Device Identifier.’ This two-part identification code is used as part of the FDA’s Unique Device Identifier System. The FDA issued its final rule on Unique Device Identifier Systems on September 24, 2013, effective December 23, 2013. The full 44-page document can be viewed on the Federal Register Website.
The idea or concept of having an identifier unique to each medical device is not a flashy new concept and has been used in other industries for many years. A UDI could be comparable to a VIN and license plates for vehicles or even social security numbers and driver’s license numbers in people. The idea is that there is a trackable piece of information that identifies individual types of medical devices.
The Two Parts of a UDI
A UDI includes two parts. One is the ‘Device Identifier’ or DI, and the other is the ‘Production Identifier’ or PI. The DI portion is the ‘Device Identifier.’ This portion of the UDI is mandatory and serves to identify the labeler and the specific model of the labeled device. Once the DI has been assigned, it is permanent and cannot be changed. Every variable of the device will require its own DI. For example, if multiple sizing options were produced for a device, then each size available would require a DI. Other variances, such as color and cosmetic or ergonomic design differences, also require separate DI numbers.
The ‘Production Identifier’ or PI and unlike the device identifier the PI identifies one of several pieces of information. I feel the best way to explain the PI’s information is to quote the FDA directly.
“a production identifier (PI), a conditional, variable portion of a UDI that identifies one or more of the following when included on the label of a device:
- the lot or batch number within which a device was manufactured;
- the serial number of a specific device;
- the expiration date of a specific device;
- the date a specific device was manufactured;
- the distinct identification code required by §1271.290(c) for a human cell, tissue, or cellular and tissue-based product (HCT/P) regulated as a device.” (FDA, UDI Basics 2015).
If a company were to produce multiple batches or lots of a device, the DI would remain the same, but the PI would differ for each batch produced.
UDI Formats
Your UDI must be provided in two separate formats. One is a plain text version that is simply an alphanumeric code that correlates with the information that it is trying to convey. This is a DI/PI code that must be labeled on the packaging of your medical device or, in some cases, on the device itself.
A second format is a form that is AIDC compatible. AIDC stands for ‘Automatic Identification and Data Capture’. AIDC collects your information without having to enter all of your data manually. Generally, this is some type of barcode or QR code.
You can see examples of AIDC technology in our daily lives. Some of the most common examples are barcodes, as mentioned above, and magnetic strips and chips as we see in our credit and debit cards. RFID, optical scanners, and other biometrics are also included as some of the less common AIDC methods.
For more information on AIDC technology in general, you can follow the 3rd party website.
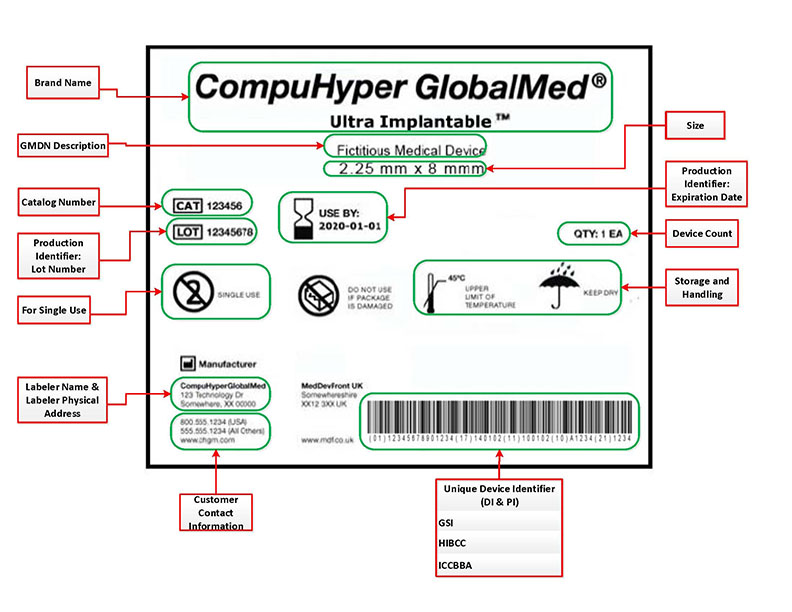
(Picture referenced from FDA website)
Where are UDI’s located?
Your UDI should be located on your device label. This is a general rule, but the FDA has multiple exemptions and alternatives based on the device use and classification. The UDI’s are required to be directly marked on the devices themselves should they be intended to be used more than once and be reprocessed before each use.
If you are writing a UDI procedure for your company, double-check that your device does not fall under any of the FDA’s exceptions.
An example of one of the exceptions that may apply to your device is, “If your device is Class I, you may use a Universal Product Code (UPC) to serve as the UDI on the device label and package. In addition, the UDI on your class I devices is not required to include a PI.” (FDA, Small Entity Compliance Guide, 2014).
Packaging Levels for UDI
Each ‘package level’ also requires a new DI. For example, if your medical device were an insulin syringe that you sold in packages of 10 and bulk in packages of 100, each would need an individual ‘Device Identifier’. This does not mean that each package of 10 or 100 needs its own DI. These are not a lot or batch numbers. These numbers are for the user’s information, so shipping materials such as pallets and shrink wrap do not require DI/PI labeling. However, different models or any substantial updates to the medical device will need its DI.
As long as your syringe is only sold as an individual syringe, the UDI and labeling are compliant. As soon as an additional packaging level is introduced, an additional UDI is required. Using the same syringe example, if the syringes are also sold in packages that contain five of your already labeled medical devices, that package needs its UDI number. Another UDI would be required if the syringes were sold in packages of ten, twenty-five, or fifty. Every level of packaging that the device is sold in requires a UDI.
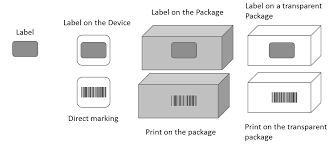
What is not considered an additional packaging level? Measures to protect your products during shipping are not considered additional packaging levels. This includes palletizing and wrapping your products to protect them from damage during shipping. Pallets, shipping containers, and trailers do not require a UDI.
Updated Products and UDI’s
UDI’s are specific to individual models of products and devices. As each packaging level or product variance, such as size offered, requires a UDI, so does each device change and upgrade. Say you launched your device 2 years ago and, based on consumer feedback, decided to make some changes to your device. Your device’s new version is no longer the same as the one that had the previous UDI issued to it.
You would now need a UDI for the essentially ‘new product.’ You must also address the same compliance requirements for packaging levels and variances as you did with the original product. As you update your product, be aware that you may need to update your UDI.
UDI date format requirements
The date format on device labels should be in the ‘International Standard’, which consists of Year-Month-Day as opposed to what would normally be seen in the United States, which is Month-Day-Year. For example, the date for April 18, 2018, would need to be written 2018-04-18.
This format would need to be used on your labeling for things such as your product’s or device’s manufacture and expiration date.
For UDI labels, the compliance date for implementing the International Date Standard will be the same as the compliance dates for UDI/AIDC.
Compliance Dates for Class I and Unclassified Devices.
Below is the FDA’s UDI Compliance Dates Table.
To extend the compliance dates for lower-risk medical devices, the FDA plans to issue a guidance document to provide an enforcement discretion policy for labeling, GUDID data submission, standard date formatting, and direct mark requirements for class I and unclassified devices, as indicated in Figure 1 below. This enforcement discretion policy would not apply to class I or unclassified implantable, life-supporting, or life-sustaining devices1 because labelers of these devices must already comply with UDI requirements.
| Type of Device | Label (21 CFR 801.20), GUDID Submission (21 CFR Part 830, subpart E), and Standard Date Format (21 CFR 801.18) Requirements | Direct Mark (21 CFR 801.45) Requirements |
| Class 1 devices2 | September 24, 2020 | September 24, 2022 |
| Unclassified devices | September 24, 2020 | September 24, 2022 |
Figure 1
1 For implantable, life-supporting or life-sustaining devices of all classes, the compliance date for all UDI requirements and the standard date format requirement (21 CFR 801.18) was September 24, 2015.
2 Class I CGMP-exempt devices are excepted from UDI requirements. 21 CFR 801.30(a)(2)
For more information, see “Letter to Device Labelers on UDI Compliance Dates for Class I and Unclassified Devices – June 2, 2017”.
Compliance Dates Established by FDA in Conjunction with UDI Final Rule
| Compliance Date | Requirement |
| 1 year after publication of the final rule (September 24, 2014) | The labels and packages of class III medical devices and devices licensed under the Public Health Service Act (PHS Act) must bear a UDI. § 801.20. Dates on the labels of these devices must be formatted as required by § 801.18. Data for these devices must be submitted to the GUDID database. § 830.300. A 1-year extension of this compliance date may be requested under § 801.55; such a request must be submitted no later than June 23, 2014. Class III stand-alone software must provide its UDI as required by § 801.50(b). |
| 2 years after publication of the final rule (September 24, 2015) | The labels and packages of implantable, life-supporting, and life-sustaining devices must bear a UDI. § 801.20. Dates on the labels of these devices must be formatted as required by § 801.18. |
| A device that is a life-supporting or life-sustaining device that is required to be labeled with a UDI must a bear UDI as a permanent marking on the device itself if the device is intended to be used more than once and intended to be reprocessed before each use. § 801.45. Stand-alone software that is a life-supporting or life-sustaining device must provide its UDI as required by § 801.50(b). |
|
| Data for implantable, life-supporting, and life-sustaining devices that are required to be labeled with a UDI must be submitted to the GUDID database. § 830.300. | |
| 3 years after publication of the final rule (September 24, 2016) | Class III devices required to be labeled with a UDI must bear a UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. § 801.45. |
| The labels and packages of class II medical devices must bear a UDI. § 801.20. Dates on the labels of these devices must be formatted as required by § 801.18. Class II stand-alone software must provide its UDI as required by § 801.50(b). |
|
| Data for class II devices that are required to be labeled with a UDI must be submitted to the GUDID database. § 830.300. | |
| 5 years after publication of the final rule (September 24, 2018) | A class II device that is required to be labeled with a UDI must bear a UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. § 801.45. |
| The labels and packages of class I medical devices and devices that have not been classified into class I, class II, or class III must bear a UDI. § 801.20. Dates on the labels of all devices, including devices that have been excepted from UDI labeling requirements, must be formatted as required by § 801.18. |
|
| Data for class I devices and devices that have not been classified into class I, class II, or class III that are required to be labeled with a UDI must be submitted to the GUDID database. § 830.300. Class I stand-alone software must provide its UDI as required by § 801.50(b). |
|
| 7 years after publication of the final rule (September 24, 2020) | Class I devices, and devices that have not been classified into class I, class II, or class III that are required to be labeled with a UDI, must a bear UDI as a permanent marking on the device itself if the device is a device intended to be used more than once and intended to be reprocessed before each use. § 801.45. |
| Compliance dates for all other provisions of the final rule. Except for the provisions listed above, FDA requires full compliance with the final rule as of the effective date that applies to the provision. | |
UDI Quality System Requirements
To comply with both Part 803.22 Medical Device Reporting and 820.198 Quality System Regulation, the documentation of UDI numbers included on device labeling is either required specifically or applicable to fulfill specific documentation and reporting requirements.
CFR 21 Chapter I Sub Chapter H Medical Devices Part 803.33 Medical Device Reporting
“(a) You must submit to us an annual report on Form FDA 3419. You must submit an annual report by January 1, of each year. You may obtain this form from the following sources:
(iv) Product model, catalog, serial, and lot number and unique device identifier (UDI) that appears on the device label or on the device package.”
(To view in full visit the regulation website)
For handling complaints as part of your quality system, inclusion of the UDI in your record of investigation is a specifically listed portion of device identifications and control numbers needed for reporting and record keeping.
Quality System Regulation Sub Part M Records 820.198
“(e) When an investigation is made under this section, a record of the investigation shall be maintained by the formally designated unit identified in paragraph (a) of this section. The record of investigation shall include:
(1) The name of the device;
(2) The date the complaint was received;
(3) Any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used;”
(To view in full view the regulation website)
UDI Issuing Agencies
All UDIs are required to be issued under a system operated by an FDA-accredited “Issuing Agency”. At the time of writing this, the FDA currently only has three FDA-accredited IA’s. They are GS1, HIBCC, and the ICCBBA. The UDI rule provides a process through which an agency would seek FDA accreditation. specifies the information that the applicant must provide to FDA and the criteria FDA will apply in evaluating applications.
To seek accreditation by the FDA as a UDI issuing agency, your UDI procedure must define the process outlined in the 21 CFR 830 Subpart C. This specifies the information that must be provided to the FDA as well as the FDA evaluation criteria. The FDA also asks that agencies seeking an initial accreditation contact the FDA directly at gudidsupport@fda.hhs.gov.
UDI Procedure for Labelers
Labelers are ultimately the ones that are responsible for complying with the FDA’s UDI labeling requirements. Are you a labeler? In most cases, but not always, the brand owner is typically the labeler.
The FDA defines a labeler as “(1) Any person who causes a label to be applied to a device with the intent that the device will be commercially distributed without any subsequent replacement or modification of the label; and
(2) Any person who causes the label of a device to be replaced or modified with the intent that the device will be commercially distributed without any subsequent replacement or modification of the label, except that the addition of the name of, and contact information for, a person who distributes the device, without making any other changes to the label, is not a modification for the purposes of determining whether a person is a labeler” (FDA, Webinar UDI 101)
Distributors add contact information only
A distributor may add their contact information to a label. As long as they are not altering the label in any other way. Alterations made to the label beyond this may constitute a change in who exactly is the labeler of the product.
Do Foreign device manufacturers need a UDI procedure?
UDI labeling rules apply to all medical devices sold within the United States and Europe. Therefore, even if your company is located outside the US or Europe, you will need a UDI procedure, and you must comply with the UDI regulations to distribute products in these two markets.
GUDID Requirements for your UDI Procedure
GUDID stands for Global Unique Device Identification Database. This database is a reference catalogue that is open for viewing by the public for every medical device with an ‘identifier’. This database can be accessed through AccessGUDID. Unlike submission, which requires an account, AccessGUDID may be accessed by anyone.
Under the UDI Rule, the FDA requires labelers who have medical devices that are labeled with a UDI to submit their device to the GUDID. If you are wondering if your device has such a labeler, we referenced above that the FDA considered the labeler to be “the person who causes a label to be applied to a device, or who causes the label to be modified, with the intent that the device will be introduced into interstate commerce without any subsequent replacement or modification of the label; in most instances, the labeler would be the device manufacturer, but the labeler may be a specification developer, a single-use device reprocessor, a convenience kit assembler, a repackager, or a relabeler.”
The GUDID is created with data about devices according to the compliance timeline table shared above and is published in conjunction with the UDI rule. The GUDID only contains the device identifier, which is the primary key to obtaining device information in the GUDID database. Production Identifiers are not submitted or stored in the GUDID.
References for your UDI Procedure
- FDA, (2015/05/06) UDI Basics, retrieved on 10/26/2017
- FDA, (2014/08/13) Unique Device Identification System: Small Entity Compliance Guide Guidance for Industry and Food and Drug Administration Staff, retrieved on 10/27/2017
- FDA, (2017/03/30) GUDID, retrieved on 11/01/2017
Additional Resources
- Federal Register Website
- AIDC Information from Tech Target
- Unique Device Identifier System: Frequently Asked Questions, Vol. 1
- UDI Guidance Unique Device Identification (UDI) of Medical Devices
- Global Unique Device Identification Database (GUDID) Guidance for Industry and Food and Drug Administration Staff
- Unique Device Identification System: Small Entity Compliance Guide
- UDI formats by FDA-Accredited Issuing Agency Version 1.3: January 27, 2017
- Title 21- Food and Drugs. Chapter 1- Food and Drug Administration Department of Health and Human Services. Subchapter H- Medical Devices. Part 830 Unique Device Identification. Subparts A-E.
- Unique Device Identification: Direct Marking of Devices (Issued Nov. 17th, 2017)