Auditing Design Controls
Learn how to apply the process approach when auditing design controls and discover how audit checklists fail to identify problems.
Examples of auditing design controls with an audit checklist:
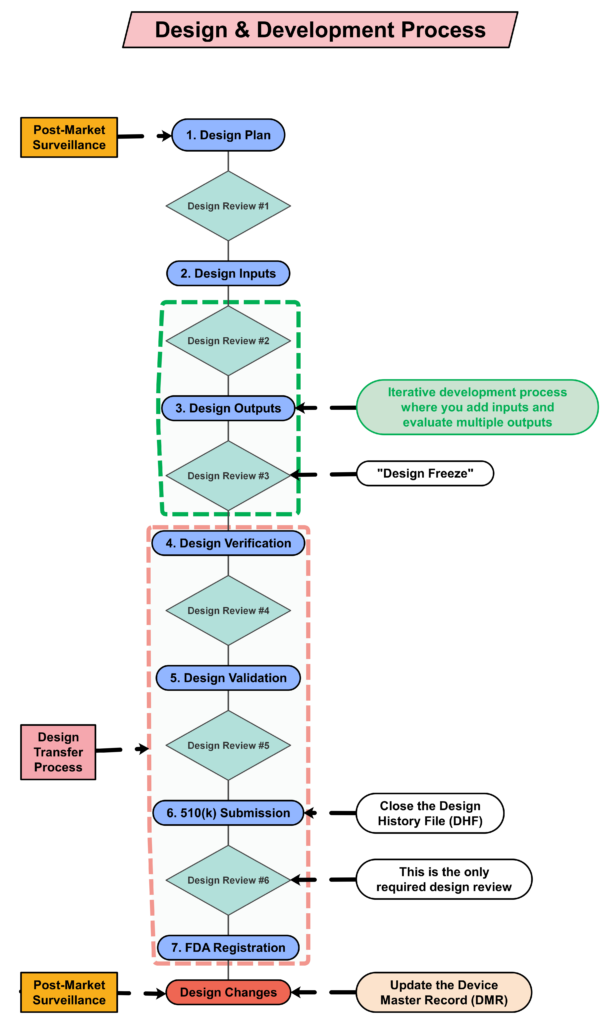
Audit checklists list each requirement in a standard or regulation. When auditing design controls, an audit checklist lists each of the ten subsections of the ISO 13485:2016 standard. For example, here are potential audit checklist questions for the first three subsections:
- Clause 7.3.1 – Which procedure(s) defines your design control process?
- Clause 7.3.2 – What is the design plan for your new product? When was the plan last updated?
- a) What are the phases of your project plan?
- b) In which phases are design reviews planned?
- c) In which project phases are verification, validation, and design transfer activities performed?
- d) Who is responsible for approval of design changes during the project? Who is responsible for updating the design plan as the project progresses?
- e) How do you demonstrate traceability between hazards, design inputs, design outputs, and testing requirements?
- f) What equipment and software do you use? What are the training requirements for your design team?
- Clause 7.3.3 – How do you document design inputs? When were the design inputs reviewed and approved?
- a) What are the performance requirements? What are the safety requirements? What are the usability requirements?
- b) What are the applicable regulatory requirements and standards?
- c) Which hazards have you identified?
- d) Which design requirements were transferred from a previous design of your device?
- e) How do you ensure that essential principles of safety and performance are met?

How can you improve the first question on the checklist?
The problem with the above questions is that they do not evaluate linkages with other processes. For example, when you ask what procedure(s) defines the design control process (e.g., SYS-008, Design Control Procedure and SYS-006, Change Control Procedure), you can also ask the revision of that procedure(s). The record associated with that document change can be used to evaluate the effectiveness of the document control process. For example, were all the job functions that reviewed and approved the previous version of the procedure represented during the review and approval of the current revision? The procedure itself can also be reviewed to make sure that it includes the appropriate elements for a procedure (e.g., scope, references, roles/responsibilities, description of the process, revision history, etc.). This approach to verifying the effectiveness of the document control process can be applied to every procedure within every process.
What are the problems with using an audit checklist?
An audit checklist is always based on the same regulation or standard. Therefore, when establishing a quality system, using an audit checklist is ideal preparation for the initial certification audit. However, if internal auditors ask the same questions during every audit, then auditees begin to anticipate the questions. Anything not included in the audit checklist may be overlooked. For example, when auditing design controls, there are no requirements for supplier controls mentioned in Clause 7.3. Therefore, an audit checklist will not include any questions about the qualification of suppliers that support design and development (e.g., software developers), as those requirements are already addressed in Clause 7.4.2 (i.e., purchasing information).
Another problem with using audit checklists is that auditors may rely too heavily on the checklist as a crutch. Auditors are supposed to plan the audit agenda based on the importance of areas and the results of previous audits. If the auditor relies solely on the checklist, each clause is assigned equal importance — regardless of its importance or the results of previous audits. Auditors also need to verify compliance with all applicable standards. Creating a checklist for risk management (i.e., ISO 14971), software lifecycle management (i.e., IEC 62304), usability engineering (i.e., IEC 62366), and information technology security management (i.e., ISO 27001) would be extremely time-consuming, and auditors would not be able to complete all of the checklist questions. Auditors require a more efficient method to assess the effectiveness of a process and verify compliance with requirements.
Basics of the process approach to auditing
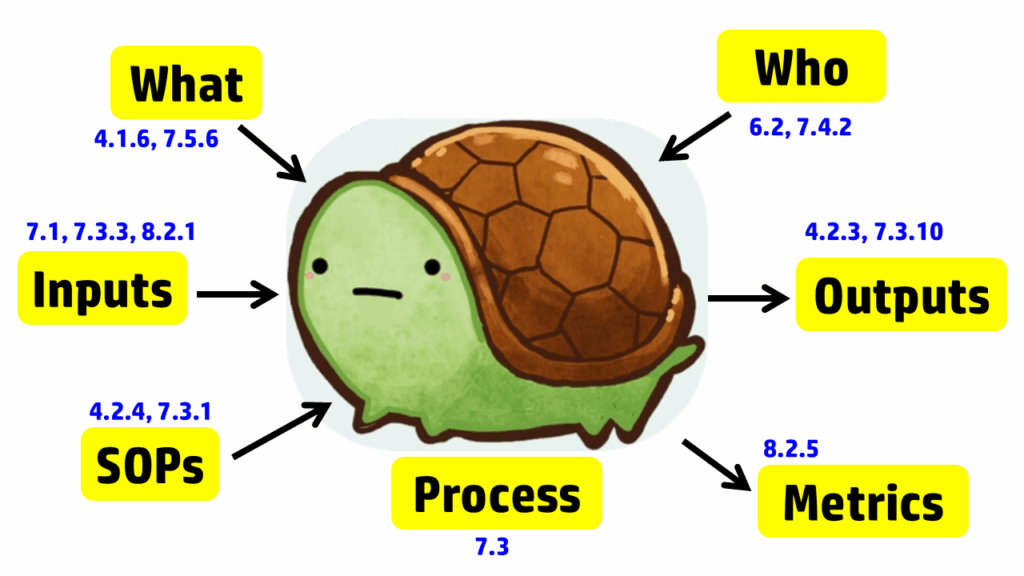
The process approach to auditing is different. Instead of creating a checklist that is specific to the requirements for each process in the standard, the process approach relies on asking seven basic questions and then following the audit trails presented by the answers to those questions. The tool we use to help us remember the seven questions is a “turtle diagram.” The shape of the turtle has seven elements:
- body (description of the processes)
- head (inputs to the process)
- tail (outputs from the process)
- leg #1 (what equipment and software is required)
- leg #2 (who performs the process)
- leg #3 (what procedures and forms are used in the process)
- leg #4 (which metrics are used to monitor the process)
Note: It’s only 7 steps. You don’t have to tattoo a turtle diagram on your arm.
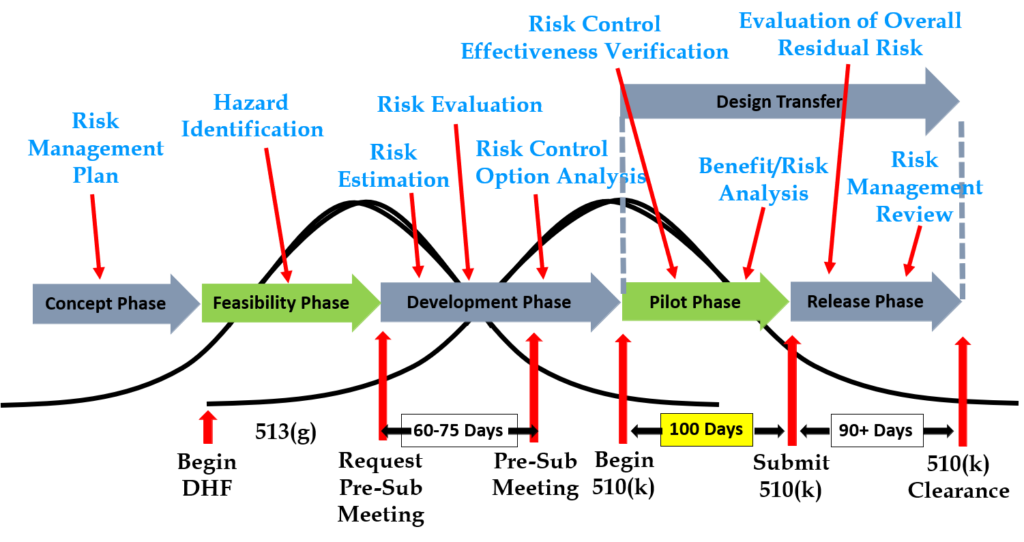
The diagram below uses the image of a turtle to remind you of the seven elements, but we added the subclauses from ISO 13485 that are related to auditing design controls. A brief summary of how these subclauses are related is provided in the video above; more details on each part of the turtle diagram are provided below, specifically for design controls.
Step 1 – Describe the process
The first step in creating a process audit is to identify the process owner and conduct an interview with them. We recommend doing this in their office, not in the conference room, for three reasons:
- Auditor effectiveness will improve if you periodically get up and walk around, because it will make you more alert.
- Conference rooms isolate auditors from daily operations, and the auditor may not gain an appreciation for where people perform their work or the proximity of the design team leader to the rest of the team.
- Auditees will be more relaxed in their office when being interviewed than they would be in a conference room.
After the process owner provides a brief description of the process, try to get answers to steps 2-7 directly from them in the same interview. Asking open-ended questions to prevent “yes/no” responses will be helpful. You also need a comprehensive understanding of the design control process before interviewing other team members or requesting design records.
Step 2 – Inputs
Even when auditors use the process approach to auditing, this part of the turtle diagram is frequently incomplete when auditing design controls. The obvious answer is to review the auditee’s approval of design inputs. This is a required record for design controls in Clause 7.3.3; however, it is not the only process input for design controls. As stated in Clause 7.3.3, “These inputs shall include…c) applicable output(s) of risk management.” Additionally, Clause 8.2.1 states, “The information gathered in the feedback process shall serve as potential input into risk management for monitoring and maintaining the product requirements as well as the product realization or improvement processes.” Therefore, both risk management and post-market feedback should be included as inputs to the design process. Using the process approach when auditing design controls will show you if the interactions between the risk management process, post-market surveillance process, and the design control process are adequate. Other inputs that should be considered for the design control process include regulatory requirements, such as:
- Common Specifications (EU)
- General Safety & Performance Requirements
- Applicable Safety and Performance Standards
- Applicable FDA Guidance
Step 3 – Outputs
Most auditors do an excellent job of covering the process outputs when auditing design controls (or any process), as the outputs typically include records, and auditors document which records they reviewed in their audit report. For the design controls process, the Design History File (DHF) (i.e., Clause 7.3.10) is the primary record sampled, and the Device Master Record (DMR) is the second most commonly sampled record. With the changes to the FDA requirements for the QMSR, auditors will be looking for a Medical Device File (i.e., Clause 4.2.3) instead; however, the records should remain the same, with just a new name. If the device is CE marked, there should also be a technical file or a technical file index.
Step 4 – What Resources
A critical part of auditing is to verify that a process is not only documented but also implemented. To implement any process, equipment, or software will likely be necessary. For the implementation of design controls, most companies utilize quality system software to manage documents and records for each design project. For example, Grand Avenue Software could be used for managing the medical device file (i.e., Clause 4.2.3), and AdaptivRisk may be used for managing the risk management file. There may also be some calibrated testing equipment that requires validation, calibration, and maintenance. Therefore, this step in the turtle diagram usually involves the following ISO 13485 clauses:
- Clause 7.5.6 – process validation
- Clause 6.3 – infrastructure (i.e., maintenance)
- Clause 7.6 – monitoring of measurement equipment (i.e., calibration)
This is typically the step of a process audit where the auditor needs to identify “what resources” are used in the process. However, only companies that have software systems for design controls have resources dedicated to Design and Development. I have indicated this in the “Turtle Diagram” presented above.
Step 5 – Who
The next step in the process approach to auditing design controls is to identify who is assigned to the design team for a design project. Sometimes companies assign large teams. In this case, the auditor should focus on the team members who must review and approve design inputs (see Clause 7.3.2) and design outputs (see Clause 7.3.4). All team members should have training records (i.e., Clause 6.2) for Design Control procedures and Risk Management procedures. However, if the device includes software and internet connectivity, some members of the design team will require additional training on specific standards and protocols. It is also necessary to outsource processes that cannot be performed by the manufacturer, such as software development, cybersecurity testing, biocompatibility testing, and EMC testing. For these outsourced processes, the company must document the supplier’s qualification and establish a written agreement with that supplier (i.e., Clause 7.4.2). Examples of agreements could be a supplier quality agreement, a consulting contract, or a signed GLP testing protocol.
Step 6 – Standard Operating Procedures (SOPs) or “How done”
Auditors using the process approach to auditing often discover ineffective processes when they expand the scope of design controls beyond the scope of the design control procedure. The design team leader will identify the design control procedure(s) and forms. However, the auditor should also request copies of the risk management procedure and other related procedures. The other procedures may have different process owners, and the design team leader may not be adequately trained in those procedures. The auditor should not read and review these procedures. Auditors never have the time to do this. Instead, ask the process owner to identify specific procedures or clauses within procedures where clauses in the ISO Standard are addressed. If the process owner knows exactly where to find what you are looking for, they’re training was effective, or they may have written the procedure(s). If the process owner has trouble locating the clauses you are requesting, spend more time sampling training records. You may also want to ask if there is another person who is more familiar with the procedure. This step of the process approach is also when you should be sampling records of document control (i.e., Clause 4.2.4).
Step 7 – Metrics
The seventh step of the turtle diagram is typically where the auditor discovers the most value-added findings. The auditor will ask the process owner to identify some metrics (i.e., Clause 8.2.5) or quality objectives (i.e., Clause 5.4.1) they are using to monitor and improve the design and development process. This is a struggle for many process owners — not just the design team leader. If any metrics are not performing up to expectations, there should be evidence of actions being taken to address this. If the process owner is not tracking metrics, you may want to review how closely the actual project schedule aligns with the design project plan. Design projects are frequently delayed because the design team either does not request quotes early enough or does not involve the supply chain manager soon enough, or both. There is also considerable benefit derived from conducting retrospective reviews at the end of design phases and at the project’s conclusion. The team will identify changes in time estimates that should be considered for future design projects or other ongoing projects.
Supplementary questions for auditing design controls
After all seven steps of the turtle diagram are complete, the process audit is not yet complete. The auditor needs to sample records and follow audit trails to ensure thoroughness. Therefore, additional records need to be sampled. We recommend sampling design changes because this is where inspectors and third-party auditors will typically focus. These external auditors will be looking for design changes that need regulatory approval and may not have been submitted for market authorization. The auditor may also sample using a risk-based approach when sampling design changes. In particular, we recommend looking for the following types of changes: 1) vendor change, 2) specification change, and 3) process change. By doing this, the audit will also cover the following clauses in ISO 13485:2016: 7.4 (purchasing), 7.3.9 (design changes), 7.5.6 (process validation), 7.1 (risk management), and 4.2.5 (control of records). If you would like to learn more about design changes, please watch our Design Changes Webinar.
Record sampling for auditing design controls
FDA inspectors and third-party auditors have similar approaches to auditing design controls. Both will begin by reviewing your procedure to verify that it includes all of the required elements of ISO 13485:2016, Clause 7.3. Next, they will sample a recent design project that was completed and request a copy of the design history file (DHF). Many design projects are behind schedule, and therefore, this is an important metric for most companies. Now that you have completed your “Turtle Diagram,” if you have more time, you can conduct interviews with team members to review their roles in the design process. You could also sample-specific Technical Files, as I indicated above. If you are performing a thorough internal audit, I recommend doing both. To learn more about using the process approach to auditing, you can register for our webinar on the topic.
Auditing Design Controls Read More »