The Waterfall Diagram was copied by the FDA from Health Canada and ISO 9001:1994, but everyone actually uses an iterative design process.

Iterative Design – What is it?
The FDA first mandated that medical device manufacturers implement design controls in 1996. Unfortunately, in 1996 the design process was described as a linear process. In reality, the development of almost every product, especially medical devices, involves an iterative design process. The V-diagram from IEC 62304 is closer to the real design control process, but even that process is oversimplified.
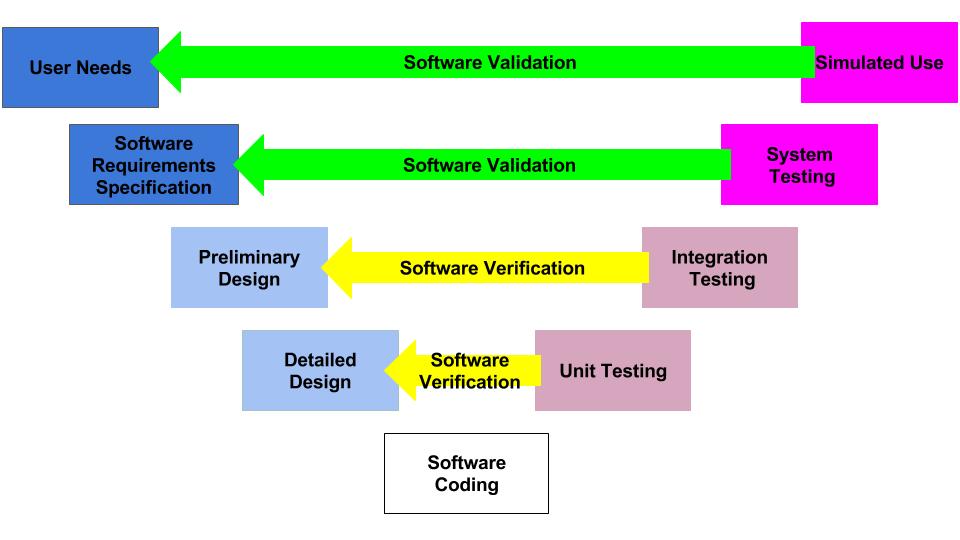
What is the design control process?
The design control process is the collection of methods used by a team of people and companies to ensure that a new medical device will meet the requirements of customers, regulators, recognized standards, and stakeholders. With so many required inputs, it is highly unlikely that a new medical device could ever be developed in a linear process. The design control process must also integrate risk management and human factors disciplines. ISO 14971:2019, the international risk management standard, requires conducting optional control analysis. Option control analysis requires evaluating multiple risk control options and selecting the best combination of risk controls for implementation. The human factors process involves formative testing where you evaluate different solutions to user interfaces, directions for use, and training. This always requires multiple revisions before the user specifications are ready to be validated in summative usability testing. Process success is verified by conducting verification and validation testing. The process ends when the team agrees that all design transfer activities are completed, and your regulatory approval is received.

Where did design controls come from?
The diagram above is called the “Application of Design Controls to Waterfall Design Process.” The FDA introduced this diagram in 1997 in the design controls guidance document. However, the original source of the diagram was Health Canada.
This diagram is one of the first slides I use for every design control course that I teach because the diagram visually displays the design control process. The design controls process, defined by Health Canada and the US FDA, is equivalent to the design and development section found in ISO 13485 and ISO 9001 (i.e., – Clause 7.3). Seven sub-clauses comprise the requirements of these ISO Standards:
- 7.3.1 – Design Planning
- 7.3.2 – Design Inputs
- 7.3.3 – Design Outputs
- 7.3.4 – Design Reviews
- 7.3.5 – Design Verification
- 7.3.6 – Design Validation
- 7.3.7 – Design Changes
In addition to the seven sub-clauses found in these ISO Standards, the FDA Quality System Regulation (QSR) also includes additional requirements in the following sub-sections of 21 CFR 820.30: a) general, h) design transfer, and J) Design History File (DHF). If you need a procedure(s) to comply with design controls, we offer two procedures:
The change control process was separated from the design controls process because it is specific to changes that occur after a device is released to the market. We also have a training on Change Control.
Free Download – Overview of the Design & Development Process
What are the phases of the Design Control Process?
Normally, we finish the design control process by launching your device in the USA because this is when you should close your Design History File (DHF). However, if you are going to expand to different markets, there is a specific order we recommend. We recommend the US market first because no quality system certification is required, and the FDA 510(k) process is easier than the CE Marking process due to the implementation of the MDR and the IVDR. The Canadian market is the second market we recommend because the Canadian Device License Application process for Health Canada is even easier than the 510(k) process for Class II devices. The Canadian market is not recommended as the first country to launch because Health Canada requires MDSAP certification for your quality system, and the Canadian market is 10% of the US market size. The European market should probably be your last market because of the high cost and long timeline for obtaining CE Marking. Each of the phases of design and development is outlined in the first column of our free download, “Overview of Regulatory Process, Medical Device Development & Quality System Planning for Start-ups.”
Which regulatory filings are required during each phase of design and development?
The second column of our free download lists the regulatory filings required by the FDA for each phase. Generally, we see companies waiting too long to have their first pre-submission meeting with the FDA or skipping the pre-submission meeting altogether. This is a strategic mistake. Pre-submission meetings are free to submit to the FDA, and the purpose of the meeting is to answer your questions. Even if you are 100% confident of the regulatory pathway, you know precisely which predicate you plan to use, and you know which verification tests you need to complete, you still have intelligent questions that you can ask the FDA. Critical questions fall into three categories: 1) selection of your test articles, 2) sample size justification, and 3) acceptance criteria. Even if you don’t have a complete testing protocol prepared for the FDA to review, you can propose a rationale for your test article (e.g., the smallest size in your product family). You can also provide a paragraph explaining the statistical justification for your sample size. You might also present a paragraph explaining the data analysis method you plan to use.
The following example illustrates how discussing the details of your testing plan with the FDA can help you avoid requests for additional information and retesting. Many of the surgical mask companies that submitted devices during the Covid-19 pandemic found that the FDA had changed the sample size requirements, and they were now requiring three non-consecutive lots with a 4% AQL sample size calculation. If the company made a lot of 50,000 masks, they would be forced to sample a large number of masks, while a lot size of 250 masks allowed the company to sample the minimum sample size of 32 masks.
If the regulatory pathway for your device is unclear, you might start with the submission of a 513(g) submission during the first phase of design and development. After you have written confirmation of the correct regulatory pathway from the FDA, you can submit a pre-submission meeting request to the FDA. If the pathway for your device is a De Novo Classification Request, you might have a preliminary pre-submission meeting to get an agreement with the FDA regarding which recognized standards should be applied as Special Controls for your device. While waiting 70+ days for your pre-submission meeting with the FDA, you can obtain quotes from testing labs and prepare draft testing protocols. After the presubmission meeting, you can submit a pre-submission supplement that includes detailed testing protocols–including your rationale for the selection of test articles, sample size justification, and the acceptance criteria.
What quality assurance documentation is required during each phase?
In addition to testing reports for your verification and validation testing, you will find many other supporting documents you also need to prepare. We refer to these supporting documents generically as “quality assurance documentation” because the documents verify that you meet specific customer and regulatory requirements. However, the documents should be prepared by the person or people responsible for that portion of your design project. For example, every device will need a user manual and draft labeling. Even though you will need someone from quality to complete a regulatory checklist to ensure that all of the required symbols and general label content are included, you will also need an electrical engineer to prepare sections of the manual with EMC labeling requirements from the FDA’s EMC guidance document. In your submission’s non-clinical performance testing section, you must include human factors documentation in addition to your summative usability testing report. For example, you will need a use specification, results of your systematic search of adverse events for use errors, a task analysis, and a Use-Related Risk Analysis (URRA). Software and cybersecurity documentation includes several documents beyond the testing reports as well.
Which procedures do you need to implement during each development phase?
The last column of our free download lists the procedures we recommend implementing during each phase of the design process. In Canada and Europe, you must complete the implementation of your entire quality system before submitting a Canadian License Application or CE Marking application. In the USA, however, you can finish implementing your quality system during the FDA review of your 510k submission or even after 510k clearance is received. The quality system requirement is that your quality system is fully implemented when you register your establishment with the FDA and begin distribution.
Pingback: Auditing Design Controls - Medical Device Academy 7 Step Process Medical Device Academy
Pingback: Implementing Design Controls Medical Device Academy