Are FDA pre-sub meetings a waste of time?
This article reviews the top reasons why other companies feel requesting FDA pre-sub meetings is a waste of time but you can’t afford to.

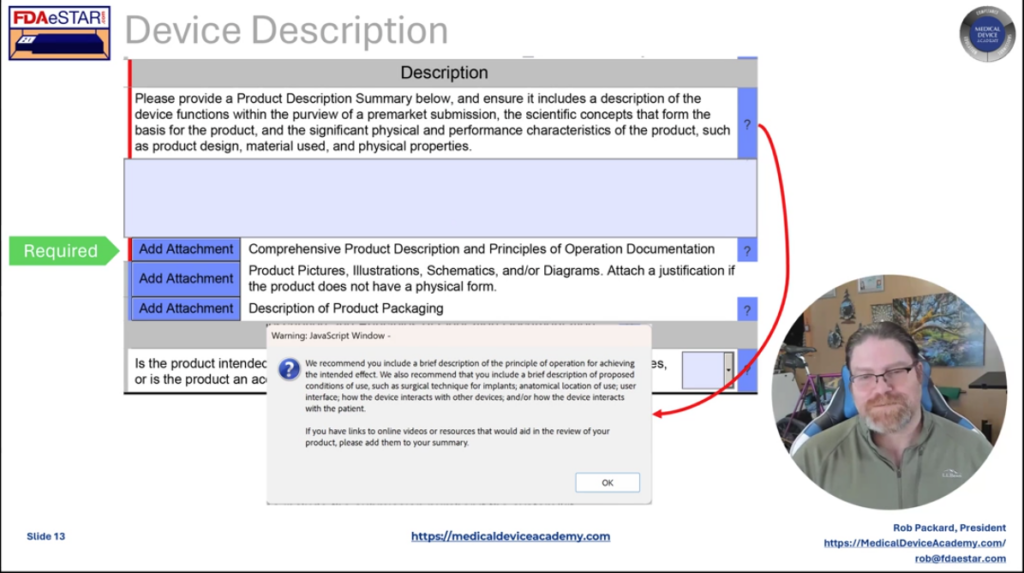
It only takes our consulting team a few hours to prepare an FDA pre-sub request using the new FDA PreSTAR. The FDA does not charge you a cent for submitting an FDA pre-sub, and a pre-sub should be part of every design plan. However, most companies are resistant to requesting FDA pre-sub meetings. In this article we review the top four reasons why companies resist submitting an FDA pre-sub request, and you will learn about the three options for the method of FDA feedback.

Our design is not finalized yet
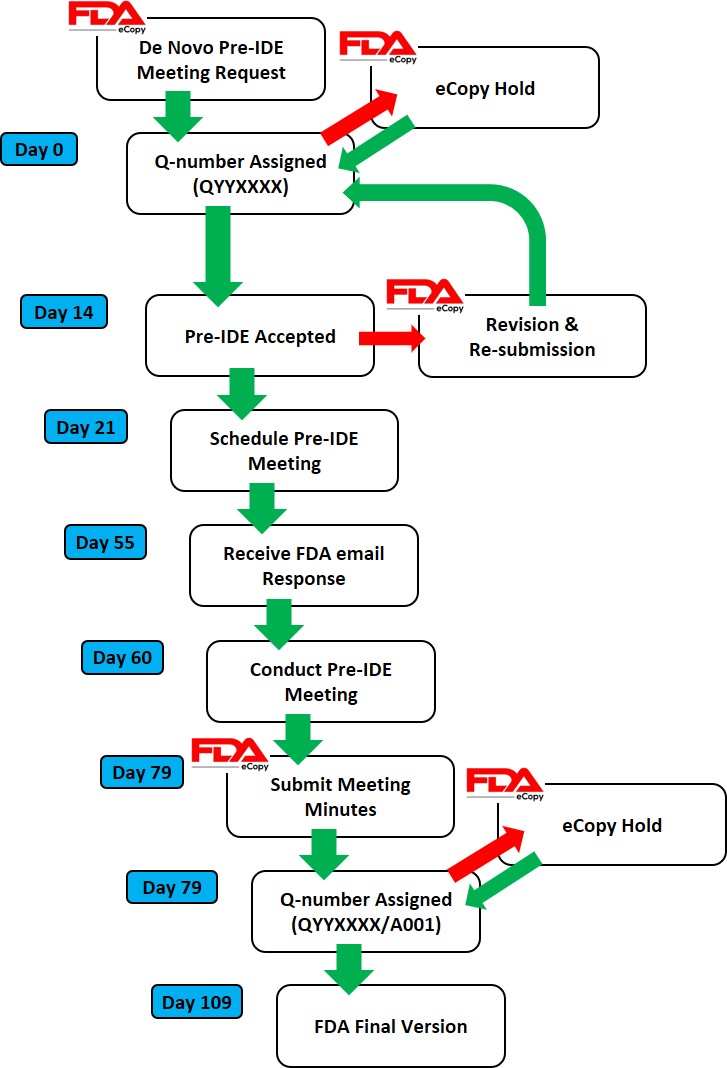
The most common reason why people delay their request for an FDA pre-sub is that they are waiting until the design is complete. This rationale is flawed because the FDA can’t review data and the FDA can’t give you advice on the design of your device. The purpose of an FDA pre-sub is to “forge a better test plan.” The FDA prefers that you submit draft test protocols with specific questions. The ideal time to submit an FDA pre-sub request is 6-9 months before you approve your design outputs (i.e., design freeze). This timing should be shortly after you approve your design inputs (i.e., essential requirements for safety and performance, standards, and stakeholder requirements). Another reason for requesting a pre-sub early is that most companies need to submit a pre-sub supplement with a revised test plan. This is especially true for biocompatibility testing, non-clinical, benchtop performance testing plans, pre-clinical animal testing, human clinical studies, and human factors testing. The revised test plan includes protocol revisions based on the feedback from the original pre-sub.

It’s too late to request FDA pre-sub meetings
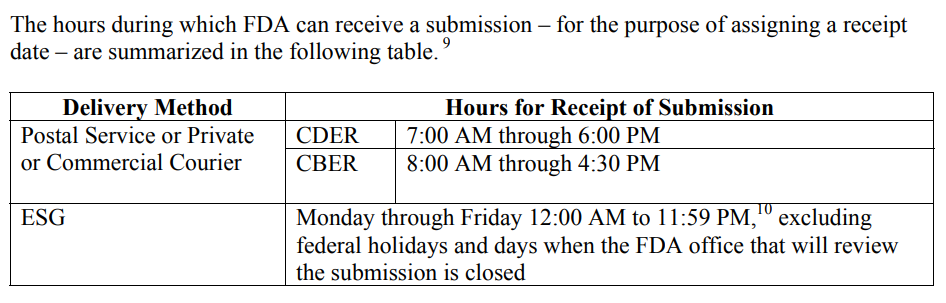
If you are less than a week away from submitting to the FDA, it is too late. The FDA target for scheduling an FDA pre-sub meeting is 70-75 days from the date your request was submitted. That’s 10-11 weeks. Most companies tell me that they plan to submit to the FDA within weeks or a couple of months, but most of the companies take nine months or longer. For example, what if your device fails EMC testing, and you have to change the design and retest for both EMC and electrical safety at an NRTL? At best, you will have an 8-week delay. If you submit a request next week, and everything goes as planned, you can always withdraw your request for the pre-sub. If you encounter a delay for any reason, suddenly, it’s not too late.
We don’t want to be bound by what the FDA says in the FDA pre-sub meeting
FDA pre-sub meetings are “non-binding.” That means that the FDA can change its mind, but it also means you don’t have to do everything the FDA says in an FDA pre-sub meeting. If you don’t ask a question about testing requirements, that doesn’t mean that the FDA does not have any testing requirements. The FDA knows what previous companies have submitted for testing better than you do, and they may be in the process of evaluating draft guidance documents. If you ask questions, you will have better insights into what the FDA expects. The most important questions are related to your rationale for why a specific testing specimen represents the “worst-case” for one of your tests. Selecting the wrong testing specimen will result in you repeating that test. Understanding FDA expectations helps you write better rationales for testing or test avoidance. You also might learn about deadlines for the implementation of new testing requirements that you might be able to avoid. Finally, you can ask the FDA about possible testing options you are considering if the FDA denies your most optimistic testing plans.
There is already a guidance document for our device
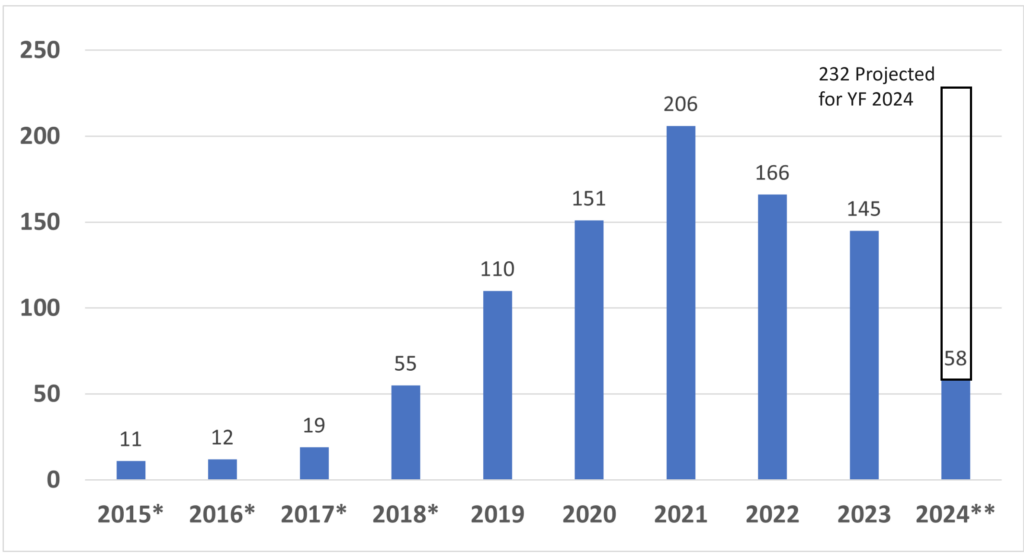
Not all device classifications have a guidance document explaining what information should be submitted in an FDA pre-market submission. However, there are almost one hundred Special Controls Guidance Documents, and for new device regulations (i.e., De Novo applications) the FDA now incorporates the special controls directly in the new regulation. Therefore, there is a good chance that the FDA published special controls as part of the regulation for your device or as a guidance document. As part of the special controls, the FDA defines what performance testing is required for your device. If you already know what testing is required, then the value in requesting FDA pre-sub meetings is diminished. But at least three other key benefits remain.
First, you can verify that the predicate you plan to use for comparative testing is not going to be a problem. Although the FDA can’t tell you which predicate to pick, the FDA can tell you if there is a problem with the predicate you have selected. This is especially important if the product is not currently registered and listed, because you may not know if the device was withdrawn from the market after it was cleared.
Second, not all testing standards are prescriptive. Many tests have testing options that require a decision. Input from the FDA may be valuable in making choices between various performance testing options. Sometimes, you forgo testing and provide a rationale instead. FDA feedback on any rationale for not doing testing is critical to prevent delays and requests for additional information later.
Third, there are many different FDA representatives who participate in FDA pre-sub meetings. The lead reviewer will invite specialists and the branch chief to the meeting. Each of these specialists can answer questions during a pre-submission meeting that they are not able to answer during the actual review process. You also have the opportunity to get feedback from the branch chief–who has insight from all the previous devices that were cleared with your product classification. Your lead reviewer is not likely to be as experienced as the branch chief, and may only have been working at the FDA for months. Your request for the 510k pre-sub meeting will help an inexperienced lead reviewer as much as it will help your company.
Which method of FDA pre-sub feedback should you request?
The FDA offers three different options for the method of feedback in pre-sub request:
- a face-to-face meeting
- a conference call
- an email response

Feedback Option 1 – A Face-to-Face Meeting
Some executives believe that face-to-face meetings are critical in establishing relationships with people. However, you need to understand the culture of the people you are trying to build a relationship with. The FDA is an overworked bureaucracy, and government agencies have security concerns. When the FDA meets with visitors they must go to a different building and arrange for their guests to pass through security. This is more work and takes more time. To justify the extra work and time, you need a compelling reason why a face-to-face meeting with the FDA is necessary.
Traveling to the FDA will cost your team money and time that conference calls and emails will not. More importantly, you are limited to one hour for a pre-submission meeting. One hour is barely enough time to ask questions and listen to the answers. You only have minutes to introduce your company and your team and describe the product. There is no time for relationship building. The best way to impress the FDA is to 1) prepare thoroughly, 2) conduct an efficient meeting, and 3) ask smart questions.
There is one time when you should visit the FDA face-to-face–if you have a powerful demonstration and video just isn’t good enough.
Feedback Option 2 – Conference Call
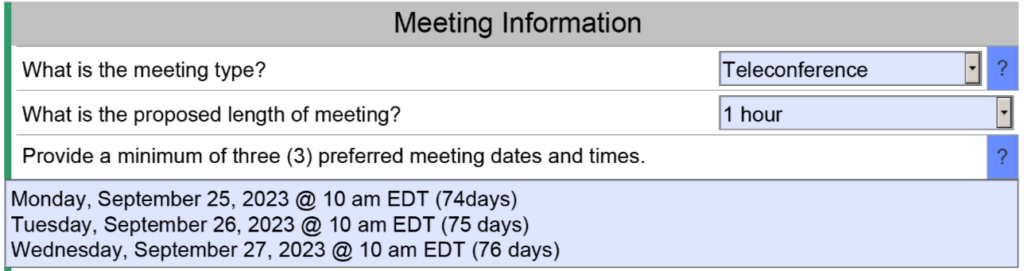
Conference calls save you time and money, but conference calls also save the FDA time and effort. You won’t personally meet people from the agency, but you can communicate information prior to the meeting and you can provide videos of simulated use for your device. Conference calls do have the advantage of allowing you to mute the call for a moment and make a comment among your team members without the agency listening as well. Whenever you are discussing a performance testing plan or a clinical study protocol with the FDA, you will probably want a conference call to enable clarification questions. The image below is an example of a request for a teleconference as the method of feedback. The FDA will still send an email response to your questions within 70 days, and you will want to submit your presentation slide deck for the teleconference at least 48 hours before the teleconference.
Feedback option 3 – Email
Email responses from the FDA are highly underrated in value. When you specify an email response, you generally receive a response to your questions sooner. You also should receive more information, because each person from the agency is able to provide an hour of their time to write detailed feedback. In a conference call, you are speaking for part of the hour, and only one person from the FDA can speak at a time. Therefore, you almost always have less feedback during conference calls and face-to-face meetings. The primary downside to email as a feedback method is that it is not interactive. In the case of submission issue review (SIR) requests (i.e., a special type of FDA pre-sub request), the FDA will only give you the option of a teleconference or email feedback–not both. Due to the challenges of scheduling teleconference, sometimes an email response can be delivered sooner and might be your best choice for an SIR request.
Learning More about FDA Pre-sub Meetings
If you would like to learn more about how to prepare the format and contents of an FDA pre-sub request, we have a four-part webinar series that will teach you how to prepare a pre-submission meeting request using the new FDA PreSTAR template.
Are FDA pre-sub meetings a waste of time? Read More »