In this article, you will learn how to spend your time during a supplier audit. We’ll teach you what is important and what you can skip.
Which suppliers need a supplier audit?
Before you start scheduling supplier audits, you should first decide which suppliers you need to audit. You are required to use a risk-based approach for supplier quality management but have specific recommendations. We recommend that you create five risk-based supplier quality categories:
Critical suppliers
Crucial suppliers
Off-the-shelf component suppliers
Service providers
Consultants
Your critical suppliers are contract sterilizers, contract manufacturers, and contract packagers. Your crucial suppliers are suppliers manufacturing custom components or subassemblies. Off-the-shelf components speak for themselves, but examples of service providers include a company doing plating and other secondary processes. The last supplier category, but not the least, is the consultant category, such as the quality system auditors you hired to do an internal audit.
Which supplier categories require a supplier audit?
The FDA regulations don’t specifically require supplier audits. However, if an inspector finds any nonconformities among your purchased components, you will need to demonstrate how you have addressed the quality issues. If the corrective actions taken are not sufficient, you will need to conduct supplier audits as part of your corrective action plan or effectiveness check. Other countries have different expectations with regard to supplier auditing, but the most common supplier categories that you will be conducting a supplier audit of are “critical suppliers” and “crucial suppliers.” These two supplier categories are also the two supplier categories that you will need to make sure are prepared and willing to accommodate unannounced audits by Notified Bodies. Click on the image below if you would like to read the requirements for audits conducted by Notified Bodies.
What is the purpose of a supplier audit?
When you attend a lead auditor course, the focus is on quality system auditing. However, when you perform a supplier audit—the quality system is not the focus. The focus of a supplier audit can fall into two primary categories: 1) qualifying the supplier or 2) re-evaluating the supplier.
Suppliers are not required to have a registered quality system or ISO 13485 certification. Therefore, many of the things that an auditor might learn about audit agendas in a lead auditor course just don’t apply. However, one thing always applies: reviewing previous quality issues. When we audit internal auditing and supplier auditing programs, we find that one of the most common mistakes is the failure to close out previous nonconformities. Therefore, the second section of my audit report template is a review of prior audit findings. If you have no previous findings, ensure your audit report states that. If you are qualifying a new supplier, ensure that the new supplier doesn’t have the same problems you are having with current suppliers.
When you close the previous issues, there are two approaches. The first approach is to close previous issues at the beginning of the audit—immediately after the opening meeting. This is the most common strategy. The second approach is to close previous issues as you audit the applicable area. For example, if you have previous problems in the area of incoming inspection and maintenance records, it might make sense to close these findings when you audit these areas. The advantage of this second approach is that it ensures that the process owner is closing the previous finding and facilitates the sampling of additional records.
What has little value in the supplier audit agenda? Auditing the management review process has the least value because the supplier is not required to have a quality management system. In fact, subcontractor audits for BSI do not include management reviews, CAPAs, or internal audits—the three required areas for every quality system audit.
What are the most valuable areas to audit?
Incoming inspection, control of nonconforming materials, preservation of the product, production controls, training, and process validation are the areas we typically audit. We would like to start with the nonconforming material area and see which materials are on hold. Then, we would like to sample the incoming inspection records for those raw materials. Next, we want to see how the company is storing those raw materials—if they are accepted. We typically cover these three areas as one process approach audit. This also happens to be the process audit we like to use for training new auditors because the audit of incoming inspection results in numerous audit trails in the support process areas of document control, training, calibration, etc.
The next area we visit is the production area. For this portion of the audit, we are doing a process audit of the production process. We usually request that we schedule the audit for a time when the production area is running the product(s) of interest. A process flow chart helps plan this portion of the audit, and we will often write some notes directly on a copy of the process flow chart.
We conclude the audit with follow-up trails in the areas of 1) document control (to ensure the supplier has the most current versions of all documentation “we” provided), 2) calibration (to ensure that all measurement devices used for inspection are calibrated), and 3) training (to ensure that all personnel working on “our” product are appropriately trained).
What are the advantages and disadvantages of skipping areas?
Since we do not have to spend time on quality system issues during a supplier audit, we spend more time sampling records in the other areas. Therefore, we might sample 5-10 records in each of the above areas instead of 3-4 records. If the number of samples available to sample is small, we may even sample 100% of the records. We also have a supplier auditor tool kit to help your supplier auditor team prepare.
Did you consider confidentiality and security issues during your supplier audit?
Historically, it has always been easy to identify a missing or out-of-date confidentiality agreement during audits, but do you include this in your internal and supplier audits? The new cybersecurity requirements that the FDA released in October 2023 certainly changed what companies need to provide in a 510(k) submission, and the latest FDA eSTAR template has a lot of specific documentation that companies need to include their 510(k). If you want to learn more about the 510(k) requirements, please visit our webpage for the cybersecurity work instruction and webinar.
How will this impact your supplier audit program?
Do you include cybersecurity questions in your supplier audits?
Do your supplier quality agreements address cybersecurity?
Do you have cybersecurity testing vendors added to your approved supplier list?
Is cybersecurity embedded in your post-market surveillance activities?
Do you and your supplier have a schedule for cybersecurity retesting?
This procedure case study describes an error-proof method for procedure review and approval of quality system procedures.
My first training in procedure review
The first time I was formally trained on how to conduct a procedure review was during a lead auditor course. I thought the topic of procedure review seemed out of place, but as I audited more companies, I realized that missing regulatory requirements in a procedure are quite common. Regardless of who reviews a procedure, or how many times it is reviewed, something is always missed. Unfortunately, a desktop audit of procedures is not an effective corrective action or verification method. Auditing procedures is an ineffective method for reviewing procedures because audits are limited by sampling.
A better approach to procedure review than auditing
Instead of random sampling, a systematic review of 100% of regulatory requirements is needed to ensure that none of the regulatory requirements are accidentally omitted. Systematically reviewing regulatory requirements for each country your company is selling in is tedious at best. You need a tool to make the reviewing process error-proof and straightforward. You also need each procedure reviewer to have a defined function to eliminate the duplication of work.
Procedure reviewer and approver roles
There are 3-5 reviewers of procedures in most companies. Some companies make the mistake of having as many as 8-10 reviewers of procedures, but more is not better. There are four primary roles for procedure review, but you could have as few as two people approving most procedures:
process owner (must review and approve)
quality management (must review and approve)
regulatory (must review, but optional approver)
independent (optional review, but not an approver)
You are not required to have all four of these reviewer roles, but including these four roles in your document control process is a best practice. Differentiating between reviewers and approvers should also be considered in your document control procedure. The only documents we recommend top management be a reviewer and approver of are:
Quality Policy
Risk Management Policy
Quality Manual
Management Review Procedure
The reason for top management reviewing these four documents is because top management has a regulatory responsibility related to each of these documents.
Process owner role
The process owner is the owner of the procedure for that process. Therefore, the process owner needs to approve that procedure. It would make no sense to own a process without the ability to approve changes. The process owner may also be the procedure author, but we don’t recommend it. Editing someone else’s work is more effective than editing your own work. Instead, we recommend that the process owner delegate the responsibility for writing and updating procedures to a subordinate who performs the procedure. Then, the process owner is responsible for reviewing and approving the procedure.
Quality management role
The quality management person needs responsibility for reviewing and approving all procedures because this person is responsible for the entire quality system. They need to make sure the procedure is accurate in the context of the entire quality system. The quality management person is the best person to review interactions with other processes. For example, the management review process has twelve required inputs (i.e., ISO 13485, Clause 5.6.2A-L). Each of those inputs comes from another process and procedure. It is essential to ensure that if you are reviewing the complaint handling procedure, somewhere in that procedure, it should state that the monitoring and measuring of complaint trends should be input into the management review process.
Regulatory role
Usually, the regulatory person is responsible for verifying that a procedure meets 100% of the regulatory requirements. This person should verify that the scope of the procedure identifies the relevant markets. If there are references to documents of external origin, the regulatory person should verify that these references are accurate. The best way to do this is by performing a gap analysis. Sometimes the quality management role and the regulatory role are combined in a small company, but larger companies will keep these roles separate. Just because the regulatory person performs a gap analysis as a reviewer, that doesn’t automatically translate to the need for approval of the procedure. We recommend making the decision on whether a regulatory person should approve a procedure based on whether the procedure has specific regulatory requirements (e.g., annual registration or regulatory reporting).
Independent reviewer role
Finally, the independent reviewer is looking for two things:
Does the procedure make sense–to someone who performs the procedure (if that person was not the author); and to an external auditor, such as a certification body (internal auditors can fill this role)?
Are there typos, spelling, or grammar mistakes?
The independent reviewer does not need to be a manager. It needs to be someone who writes well. Editing is tedious, but apparent mistakes in spelling or grammar prompt auditors to review procedures more carefully. If available, we recommend asking an internal auditor to be the independent reviewer. Depending upon the experience of the independent reviewer with regard to performing a gap analysis, the person with regulatory responsibility may delegate the task of gap analysis to independent reviewers. This role can also be satisfied by a consultant with technical writing ability. Medical Device Academy’s resident expert at this is Matthew Walker.
Procedure case study – The most common auditor findings
The two most common reasons for audit findings are:
the procedure is not being followed, and
a regulatory requirement is missing from your procedure.
Not following the procedure
The first problem is the most common reason for audit nonconformity, as companies include requirements in the procedure that are not regulatory requirements. Auditors look for objective requirements to audit. Therefore, if you include objective requirements in your procedure an auditor is more likely to select those requirements to sample than subjective requirements–even if the requirement is not a regulatory requirement. This is one of the reasons we recommend having processing owners review and edit procedures. If you purchase a procedure, it’s important for the person who will be performing the procedure to carefully review the procedure to ensure it matches how they intend to perform that process. If it’s a manufacturing procedure, we recommend training personnel with a draft procedure and handing out red pens. That also dramatically reduces complaints from the people who do the work.
Regulatory requirements missing
For regulatory requirements, your regulatory reviewer needs to create a checklist that includes 100% of the requirements for that procedure. This approach is called a gap analysis. The model for gap analysis documentation we like to follow is the General Safety and Performance Requirement (GSPR) Checklist used for technical documentation (i.e., for CE Marking). There are 23 GSPRs in the MDR and 20 GSPRs in the IVDR. Most of the GSPR requirements have multiple subparts. The regulatory person who completes the GSPR Checklist must indicate the following information next to the applicable requirement in the checklist table:
yes, the requirement applicable or justification if it’s not applicable
a reference to any applicable standards
a cross-reference to the record where evidence of meeting the requirement can be found (e.g., the risk management file)
Regulatory personnel can revise this approach slightly by doing the following for the review of procedures:
yes, the requirement applicable or justification if it’s not applicable
a reference to the applicable specific sub-clause in a Standard or a regulation
a cross-reference to the subsection of the procedure where evidence of meeting the requirement can be found (e.g., section 5.1 of the SYS-003)
Procedure Case Study of the Management Review Procedure (SYS-003)
In Medical Device Academy’s Management Review Procedure, Section 8 is the “procedure section.” Sub-section 8.3 of the procedure lists all the required inputs for a Management Review meeting. Next to each input, we included a cross-reference to the sub-clause in ISO 13485:2016 for the Management Review input.
There is also a requirement in ISO 13485:2016 for conducting Management Reviews at scheduled intervals. This requirement is met by sub-section 8.1 of the Management Review procedure. We used the same approach to identify and cross-reference to this requirement.
Teaching auditors by performing your own procedure case study
Now, when we teach our Lead Auditor Course, we ask attendees to split into small groups to review a procedure–one procedure for each group. In one of the companies where we did this, each of the four teams found a regulatory requirement that was missing from the procedures they were reviewing. All four procedures the teams selected were already reviewed, approved, and currently in use at the time of the auditor training. The four teams created their own procedure case study to demonstrate the importance of reviewing procedures for regulatory requirements.
The author discusses his personal experience with quality and regulatory training and shares his secrets for better instructor engagement.
What is instructor engagement?
Instructor engagement is a term that describes actions taken by the instructor to involve their class, whether the training is on-line or in person. Instructor engagement includes six basic elements:
Training content and format
Verbal communication
Non-verbal communication
Training environment
Audience
Audience involvement
Any one of the above six elements can ruin a training class, but a great instructor can compensate for weaknesses in any one area by taking advantage of the other elements.
To be successful, you need to hook your audience in the first ten seconds
It is crucial to engage your audience in the first ten seconds. When the audience is live, if you don’t engage them immediately they will find something on their phone to distract them. If the audience is on-line, they will swipe to the next video in their feed. Toastmasters suggests beginning your presentation by using one of five methods for hooking your audience:
Tell a story
Make a bold statement
Ask a question
Get the audience to laugh
Ask the audience to visualize something
Consequences of poor engagement
If an instructor does not engage students, the best case scenario is that the training will not be effective. In other words, the class will not learn the material being taught or they will retain the information for less than 24 hours. For the student, their time and money was wasted. For the instructor, they will feel exhausted at the end of the training and they will have trouble finding future training jobs.
Results of good instructor engagement
If a class is engaged in training they will learn the material, retain the information in their long-term memory, and the will recommend the instructor to other people that are interested in learning about the topic. For the student, their time and money was well spent. For the instructor, they will feel energized at the end of the training and students will come up to them at the end of the training asking for a business card and to discuss future training opportunities.
Where can you find an examples of good and bad instructor engagement?
When I first wrote a blog on this topic it was 2012 and there were very few blogs and almost no videos dedicated to quality systems or regulatory affairs. Twelve years later, almost nobody reads blogs and there are multiple competitors that publish new videos weekly. The primary channel for watching videos is YouTube, but YouTubers are simultaneously live-streaming on YouTube, LinkedIn, and Facebook. These channels are the best place to find examples of good and bad instructor engagement. You will probably have a strong opinion about the quality of the speaker in the first ten seconds, but the algorithms that guide your surfing of these platforms will automatically steer your viewing to the best videos. These videos are not considered to be the best solely on content and format. The amount of audience engagement is the biggest driver. The algorithm recommends videos based upon the percentage of audience retention, the number of comments by viewers, and the number of people that share the link to your video.
Why doesn’t Dr. Shulman’s video rank higher in the algorithm?
The video I embedded in this post has only 554 views currently, but it has been posted on YouTube almost six years. Why doesn’t this video rank higher and get recommended by YouTube to more people? Because there is no involvement of the original audience or the current YouTube audience. Despite the talented speaker and the use of video with clear PPT slides, there are no comments on YouTube. The content and format is good, and the verbal communication is good. However, RAPS has Dr. Shulman standing behind a podium at the event so the non-verbal communication is not as strong as it could be. In addition, large conferences are typically one of the weakest environments for encouraging instructor engagement with the audience. The video could receive much higher rankings if the original audience was involved in the video as well. If the question and answer session were included, that might have helped. RAPS could also improve the video’s performance by adding more details to the description of the video. Finally, the performance of the video is impacted by the number of subscribers to the RAPS YouTube channel and the engagement of those subscribers. To demonstrate this, I will add a comment to the video. That should cause a small increase in engagement and viewers.
How can you improve your internal quality system training?
Anyone can read and understand a procedure, but this is the least effective method of training people. You could also have employees watch a training video, but quality assurance and regulatory affairs are among the most boring topics on planet earth. Most of the training out there is “Blah, blah, blah…” and “Death by PowerPoint.” Instructor engagement for that type of training is poor, and it could get you fired. Don’t read your slides, don’t turn your back on the audience (or they’ll attack), and PLEASE don’t ever ask someone to read the definition of nonconformity out loud to the rest of the group. Inspire and engage the class. You need to get your audience to pay attention, ask questions, and share their own thoughts out loud. For example, instead of using a PowerPoint, try displaying the actual procedure and ask an audience member to find each of the points you are teaching in the procedure. You could even teach them a cool search tool (i.e., CTRL + F) to find the content. You might even adding a symbol to the procedure to help them find those requirements.
Nine ways to improve your own instructor engagement
If you are hiring a consultant to help you with quality and regulatory training, then you certainly want to hire an expert. However, it is more important that the speaker is engaging. Knowing this fact, you could try improving your own presentation skills to achieve higher instructor engagement for a lot less money. I’m six-foot, six inches tall, and I have a loud booming voice. My mother has red hair, and she was an opera singer. I’ve got the voice to fill any auditorium and stage presence to match. But you don’t have to be big, tall, or loud to capture the attention of your audience. Here are my top nine ways to improve instructor engagement:
Practice vocal variety
Move, don’t stay stationary
Ask the audience questions
Use anecdotes, case studies, and stories
Try using props
Take breaks
Plan a surprise
Force feed the audience legal stimulants
Give students homework
What is vocal variety, how does it impact instructor engagement?
Vocal variety is more than just the volume of the speaker. Vocal variety consists of pitch, tone, volume, and pace. Generally I speak too fast and my voice is very loud. Therefore, if I want to emphasize a point I can exercise two changes in my voice to immediately capture the attention of students: 1) speak softly, and 2) speak slowly. Another approach I have used, is to speak slowly and repeat myself, but the most dynamic way to get your audience’s attention is to stop speaking for a few seconds. Silence is powerful.
Many people struggle to understand how to vary their pitch, but they’re overthinking it. When we ask a question, we raise the pitch of our voice slightly at the end. This upward inflexion of our voice signals to listeners that we are asking a question. For example, if you repeat the last one to three words in the other person’s sentence, and you say this with an upward inflection, the listener will perceive that you are asking a question to better understand what they mean. This is much more effective than asking “what do you mean?” For example, if a student in the class says “Is it enough if we perform annual reviews?” As the instructor you can employ the technique of mirroring by asking, “Annual reviews?” This will encourage the other person to elaborate on what they meant by “annual reviews.” This technique ensures that you know what the student meant, and it gives you time to consider what they said before you respond. This is also a strategy recommended by expert negotiators.
Movement is attractive
Most of our brain power is dedicated to processing what we see–not what we hear. Therefore, listeners are more likely to notice when you move. You can jolt your audience awake simply by stepping out from behind a podium or changing your movement pattern (e.g., occasionally moving forward instead of pacing side to side). Movement also includes body language. You can modify your posture, stance, and position in front of the audience to communicate information non-verbally. You can use gestures to communicate non-verbally or you can use facial expressions. For example, if you frown and shrug your shoulders, what does that mean?
Questions are essential for instructor engagement
No matter how smooth and eloquent your voice is, nobody wants to hear only you speak. This is one of the reasons conferences have multiple speakers. However, during a single presentation you can get students to participate and present some of the information by asking them questions. There are a few techniques that help get the audience to speak more. Instead of asking a question to the group as a whole, try pointing to a specific person, ask them their name, and then ask them a question. Second, in order to “break the ice” at the end of your presentation, have some “seed questions” prepared. If you are conducting a webinar, seed questions can be read by you. In a live presentation, you can give a list of seed questions to your host or friends in the audience. Finally, you can also begin your training with a question (i.e., Toastmaster hook #3 above).
Tell a story and make a point
For each teaching point you should consider using an anecdote, case study, or a related story. The audience will want to know how the story ends, and they are more likely to remember the story. If you haven’t conducted more than 1,000 audits, traveled all over the world, or have more than 25 years of experience–don’t despair. You can always interview other people on any topic to get their stories. If you are looking for tips on how to construct a story, Toastmasters comes to my rescue again. Here’s their recommended six-part structure for the Hero’s Journey:
Setup
Inciting incident
Progressive complications
Insight
Climax and resolution
Lesson
What kind of prop can you use for quality and regulatory?
In one of the paragraphs below, I mention a simple prop that you can use for training–product samples. I did this in one of our live-streaming YouTube videos where I explained how to review medical device labeling. But you can use other things in your environment. For example, the first public speaking course I ever had was taught by a man with Polio that used crutches. He used one of his crutches as a prop to demonstrate how he looked over a fence.
How often should you take breaks?
Students cannot maintain a state of alertness and attention indefinitely. Your body naturally cycles between higher and lower alertness every 90 minutes. After 90 minutes it becomes harder to focus and you need to take a break. If you can, splitting 90 minutes into two 45-minute sessions is even better. You can also experiment with two strategies for better instructor engagement: 1) conduct a “pop quiz” after a break to make sure the audience understood the information they were just taught, and 2) don’t be afraid to adjust the breaks slightly to coincide with a change in topic. Changing topics at a break allows you to repeat the most important points three times. The first time when you introduced a topic, the second time when you have concluding remarks at the end of a topic, and a third time after the break when you make sure the audience understood the material you presented before the break.
What kind of surprise will engage your audience?
There are two strategies for using a surprise: 1) promise to surprise them in the future, or 2) don’t tell anyone until you surprise them. The first strategy works best when you are trying to get people to watch until the end of the training. This is commonly used by YouTubers to get people to watch the video until the end. Unfortunately, this backfires because we can fast-forward to the end. I’m not suggesting that you shouldn’t surprise your audience, but the surprise needs to delight your audience. You might also need to surprise and delight your audience more than once. Even then, some of your audience will still nod off and completely ignore you. When this happens, throw a Snickers bar at the offending student.
This is an essential tool for any instructor. It functions as a tool to prod sleeping students awake, is small enough to cause minimal injury when thrown, serves as an emergency food supply, and is gluten-free.
How to force-feed students legal stimulants
If legal counsel recommends against using projectiles to encourage class participation, you might also consider one of my all-time genius ideas–consuming dangerously large quantities of caffeine. I was scheduled for a two-day course in Ottawa, but the day before I needed to perform an audit in Pennsylvania. My flight was the last flight into Ottawa, which arrived at approximately 1 o’clock in the morning. My arrival was delayed an additional hour in customs by the person in front of me who was trying to smuggle an extra carton of smokes into the country. Just before 4 a.m., my taxi arrived at the Albert at Bay Suite Hotel. The class started at eight in the morning. I made it to class on time, and the excessive consumption of several pots of black coffee helped get me to lunch. Then my legs started getting a little shaky. Fortunately, there was a convenience store next door that sold my favorite chocolate–the Dark Aero bar! After four of these monstrous doses of cacao, and another pot of coffee, I could have listened to the lecture on the Canadian Medical Devices Regulations all night. The only problem is that my hands are still shaking 15 years later.
Hershey’s copied them, but the result was a mere shadow of Nestle’s greatness. Canadians know how to make junk food, tell a joke, and play hockey!
Why was the instructor engagement high in Ottawa?
Despite the physical handicap of sleep deprivation, I still learned a ton from my course in Canada. Here’s why:
The instructors were both regulatory experts that were able to share anecdotes, case studies, and stories about real-world application of the Canadian Medical Devices Regulations. One of the instructors even worked for Health Canada.
The audience was hyper-motivated to pass the course, because everyone in the class worked for a Notified Body that had sponsored them to take the course. In order to stay employed and get a raise, I needed to pass that course. If I failed the exam, I had to absorb the cost to travel back to Ottawa and retake the course in February (BRRRR!).
The instructors brought more than a dozen medical devices to the class. These props gave us something to read, touch, and ask questions about. The instructors broke us up into small teams to study the labeling and instructions for use of each device. Even students from Japan, Europe, and Australia were familiar with some of the products. This was critical because we all needed to be able to identify incorrect Canadian labeling.
The best instructor engagement tool used was humor. The instructor from Health Canada was hilarious. He had everyone laughing at his jokes for the entire course. Most of the jokes were not funny enough for a stand-up routine, but this was a mandatory regulatory course on Canadian regulations. Who would even expect a chuckle? Despite the strengths of these instructors, there is only one reason why I know the Canadian Medical Devices Regulations (CMDR), as well as I do. I use them every single week.
When students are forced to do homework, they will pay attention
After completing my CMDR training, I had to audit 162 days for BSI in 2011. Ninety percent of those 162 days were for companies that required a Canadian Medical Device License. This forced me to use the information I learned in the course. I was also consulting for companies at the same time I was auditing for BSI. Consulting clients hired me to prepare and submit the Canadian Medical Device License Applications for them. I also had to create procedures for Canadian Licensing, Incident Reporting, and Recalls. I spent another 60+ days in 2011 doing consulting, which helped me hone my knowledge of Canadian device regulations.
Teaching others will make you a guru
Most people are terrified to speak on any topic–even to a small group of coworkers. However, I believe that teaching others is the secret to becoming a guru on any topic. I was one of BSI’s instructors that taught the regulatory comparison course from 2010 to 2012, which compared the regulations of the USA, Canada, Europe, Australia, and Japan. Therefore, at least once a month, I had a classroom of 6-20 people asking me challenging questions about how to interpret and apply regulations from each of these countries to their products. I used every bit of knowledge I learned in that course in Ottawa, and I started using that knowledge immediately after the course. Peers, clients, and students challenge my knowledge of these topics every day. This is what makes you a subject matter expert. If you need to learn something about quality assurance or regulatory affairs watching a one-hour webinar, reading a blog, taking a five-day course, or shadowing another more experienced person is not enough. In the end, all of the above will get you to the level of barely competent! If you want to master any topic, you need to practice instructor engagement and use everything you learn for several years.
Data hazards occur when there is missing data, incorrect data, or a delay in data delivery to a database or user interface. This can occur with both software as a medical device (SaMD) and software in a medical device (SiMD). Missing data, incorrect data, and delays in data delivery cannot cause physical harm to a patient or user. Therefore, many medical device and IVD manufacturers state in their risk management file that their device or IVD has low risk or no risk. This is an incorrect software risk analysis because failure to meet software data or database requirements results in a hazardous situation, such as 1) an incorrect diagnosis or treatment, or 2) a delay in diagnosis or treatment. These hazardous situations can compromise the standard of care at best, or at worst, hazardous situations can result in physical harm–including death.
Where do you document these hazards?
Data hazards are documented in your software risk management file, but the data hazards are referenced in multiple documents. Usually data hazards will be referenced in a software hazard identification, the software risk analysis, software verification and validation test cases, and the software risk management report. Security risk assessments will also identify potential data hazards resulting from cybersecurity vulnerabilities that could be exploited.
How do you identify data hazards?
IEC 62304 is completely useless for the purpose of identifying data hazards. In Clause 5.2.2 of the standard for the software life-cycle process, the only examples of data definition and database requirements provided are form, fit, and function. IEC/TIR 80002-1, Guidance on the application of ISO 14971 to medical device software, is extremely useful. Specifically, in Table B1 the following potential data hazards are identified:
Mix-up of Data such as:
Associating data with the wrong patient
Associating the wrong device/instrument with a patient
Associating measurements with the wrong analyte
Loss of data resulting from events such as:
Connectivity failure or Quality of Service (QoS) issues
Incorrect data acquisition timing or sampling rates (i.e., measurement)
Capacity limitations during peak loads
Missing data fields (i.e., incorrect database configuration or database mapping)
Modification of data caused by:
Data entry errors during manual entry
Automated preventive maintenance
Reset of data
Rounding of data
Averaging of data
Patient data also presents a security risk. Access to data must be controlled for data entry, viewing, and editing. Other potential causes of data integrity issues include power loss, division by zero, overflow/underflow, floating point rounding, improper range/bounds checking, off-by-one, hardware failure, timing, and watch-dog time outs. In Table B2 the guidance provides additional examples of causes and risk control measures are recommended for each cause.
Data hazards associated with artificial intelligence (AI) and machine learning (ML)
There are also data hazards associated with AI/ML software. When an algorithm is developed there is a potential for improving the algorithm or making the algorithm worse. There is always a data bias resulting from the patient population selected for data collection and the clinical users that assign a ground truth for that data. Sometimes the data entered is subjective or qualitative data rather than objective, quantitative data. The sequence or timing of data collection can also impact the validity of data used for training an AI algorithm. AAMI CR 34971:2023 is a Guide on the Application of ISO 14971 to Machine Learning and Artificial Intelligence. That guidance identifies additional hazards associated with data and databases.
How are these hazards addressed in software requirements?
In IEC 62304, Clause 5.2.2, lists the content for twelve different software requirements (i.e., items A-L):
a) functional and capability requirements;
b) SOFTWARE SYSTEM inputs and outputs;
c) interfaces between the SOFTWARE SYSTEM and other SYSTEMS;
d) software-driven alarms, warnings, and operator messages;
e) SECURITY requirements;
f) user interface requirements implemented by software;
g) data definition and database requirements;
h) installation and acceptance requirements of the delivered MEDICAL DEVICE SOFTWARE at the operation and maintenance site or sites;
i) requirements related to methods of operation and maintenance;
j) requirements related to IT-network aspects;
k) user maintenance requirements; and
l) regulatory requirements.
These software requirements may overlap, because any specific cause of failure can result in multiple types of software hazards. For example, a loss of connectivity can result in mix-up of data, incomplete data, or modification of the data. Therefore, to ensure your software design is safe, you must carefully analyze software risks and evaluate as many test cases as you can to verify effectiveness of the software risk controls.
What is the best way to analyze data risks?
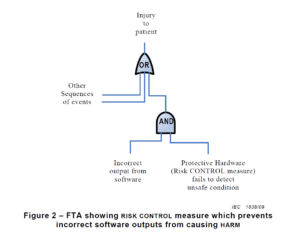
There are multiple risk analysis tools available to device and IVD manufacturers (e.g., preliminary hazard analysis, failure modes and effects analysis, and fault-tree analysis). Using a design failure modes and effects analysis (i.e., dFMEA) is the most common risk analysis tool, but a dFMEA is not the best tool for software risk analysis. There are two reasons for this. First, the dFMEA is a bottom-up approach that assumes you know all of the software functions that are needed–but you won’t. Second, the dFMEA will have multiple rows of effects for each failure mode because each cause of software failure can overlap with multiple software functions. Therefore, the best way to analyze data risks is a fault-tree analysis (i.e., FTA). The FTA is the best tool for analysis of software data hazards because you only need three fault trees: 1) Mix-up of data, 2) Loss of data, and 3) Modification of data. In each of these FTAs, all of the potential causes of software failure will be identified in the branches of the fault tree. Analyzing the fault tree structure, specifically the position of OR gates, can assist in software design. OR logic gates that can result in critical failures need additional software risk controls to prevent a single cause of software failure from creating a serious hazardous situation.
Copied from Section 6.2.1.5 from AAMI TIR 80002-1:2009
How to build a fault tree
The first step of your software risk analysis should be to identify data hazards. Once you identify the data hazards, you can build a fault tree for each of the three possible software failures: mix-up of data, 2) incomplete data, and 3) modification of data. Each data hazards can cause multiple software failure modes, but the type of logic gate will determine the outcome of that data hazard. An OR gate will result in software failures if there is just one hazardous event, while an AND gate requires at least two hazardous events to occur before the software failure will occur. The position of the OR/AND gate also impacts the potential for software failures.
GSPRs are the General Safety and Performance Requirements for CE Marking of medical devices and IVDs in Annex I of the EU MDR and IVDR.
What are the GSPRs?
General Safety and Performance Requirements (GSPRs) are the requirements for safety and performance specified in Annex I of the EU MDR and EU IVDR. GSPRs are divided into Chapter I (i.e., – Sections 1-9 of the MDR and Sections 1-8 of the IVDR are the General requirements), Chapter II (i.e., – Sections 10-22 of the MDR and Sections 9-19 of the IVD are the Requirements regarding design and manufacture), and Chapter III (i.e., Section 23 of the MDR and Section 20 of the IVDR are the requirements regarding the information supplied with the device or IVD). All devices must meet the requirement of Chapter I and Chapter III, but the applicability of Chapter II depends upon the technological characteristics of the device or IVD.
Where do the GSPRs go in your technical documentation?
When a Notified Body reviews your technical documentation, they expect you to provide either a complete technical file or technical file index that is organized in accordance with Annex II of the MDR or IVDR. Section 4 of Annex II is labeled “General Safety and Performance Requirements.” This section is where your GSPR checklist should be located in the technical file or technical file index. Generally a GSPR checklist is considered the best way to document these requirements. The checklist should should provided traceability to each specific requirement and the following elements:
the applicability or non-applicability of each requirement; justifications shall be documented for non-applicability
the method or methods used to demonstrate conformity with each requirement
the harmonized standards (i.e., EN standard) and/or Common Specifications (CS) applied
identification of controlled documents that provide evidence of conformity with the harmonized standards or CS
What are the subparts of each chapter in the GSPRs?
Chapter I
The general requirements for safety and performance (i.e., Chapter I in Section 1-9 of the MDR and Section 1-8 of the IVDR) are primarily focused on risk management requirements. These first few sections state that the manufacturer must ensure that the device or IVD is safe, effective, and does not compromise the clinical condition or safety of patients or users. The manufacturer must take into account the generally acknowledged state of the art. Risks must be reduced as far as possible without adversely affecting the benefit-risk ratio. The manufacturer must implement a risk management system. Risks associated with use errors shall be eliminated or reduced as far as possible. The characteristics of performance shall not be adversely affected the conditions of use, transport and storage during the lifetime of the device or IVD. Finally, all residual risks shall be minimized and be acceptable when weighted against the benefits to the patients and/or user arising from the intended use during normal conditions of use.
Chapter II of the MDR
This Chapter of the GSPRs is organized into the following subsections of the MDR:
10 – Performance characteristics
11 – Chemical, physical and biological properties
12 – Infection and microbial contamination
13 – Devices incorporating materials of biological origin
14 – Construction of devices and interaction with their environment
15 – Devices with a diagnostic or measuring function
16 – Protection against radiation
17 – Electronic programmable systems
18 – Active devices and devices connected to them
19 – Particular requirements for active implantable devices
20 – Protection against mechanical and thermal risks
21 – Protection against the risks posed to the patient or user by devices supplying energy or substances
22 – Protection against the risks posed by medical devices intended by the manufacturer for use by lay persons
Chapter II of the IVDR
This Chapter of the GSPRs is organized into the following subsections of the IVDR:
9 – Performance characteristics
10 – Chemical, physical and biological properties
11 – Infection and microbial contamination
12 – Devices incorporating materials of biological origin
13 – Construction of devices and interaction with their environment
14 – Devices with a measuring function
15 – Protection against radiation
16 – Electronic programmable systems
17 – Devices connected to or equipped with an energy source
18 – Protection against mechanical and thermal risks
19 – Protection against the risks posed by devices intended for self-testing or near-patient testing
Chapter III
Chapter III is divided into four subparts. Section 23.1 of the MDR and section 20.1 of the IVDR are the general requirements for information provided by the manufacturer (i.e., labeling). The recommended harmonized standard is EN ISO 20417:2021. Section 23.2 of the MDR and section 20.2 of the IVDR include the labeling requirements. Section 23.3 of the MDR and section 20.3 of the IVDR include requirements for information on the packaging which maintains the sterile condition of a device or IVD (i.e., label on the sterile barrier packaging). Finally, Section 23.4 of the MDR and section 20.4 of the IVDR include requirements for the Instructions for Use (i.e., IFU, Directions for Use, or User Manual).
Completing your checklist
Completing the GSPR Checklist would be easy if there were only 20-23 requirements, but most of the requirements have multiple requirements. For example, GSPR 14 of the MDR has 7 subparts, 18 of the MDR has 8 subparts, and labeling requirements are six pages long. Each subpart must be addressed when you complete the columns of the checklist. If any of the parts or subparts do not apply to your device, you need to provide a justification. When you write your justification for the non-applicability of a GSPR, you need to be careful to provide a justification for each subpart of the requirement–even if the subpart is not separately identified by a letter or number.
Download our Checklist
If you need a template for creating your own GSPR checklist, you can download our template by filling in the form below:
How do you address differences from the Essential Principles of Safety and Performance?
Health Canada also identifies Essential Principles for Safety and Effectiveness in Sections 10-20 of the Canadian Medical Device Regulations (i.e., SOR 98/282) that is similar to the European GSPRs, and Australia has a similar Essential Principles Checklist document with only a few minor differences. The Global Harmonized Task Force (GHTF) created an earlier version in 2005, but the International Medical Device Regulators Forum (IMDRF) released a newer version in 2024. Health Canada will typically accept your GSPR checklist developed for CE Marking, but a gap analysis should be performed against the Australian Regulations.
CE Marking auditors may ask if you assessed the difference between the ISO version of a standard and the EN standard. Is there a difference?
What is an EN standard?
European Standards are technical standards ratified by one of the three European standards organizations: CEN, CENELEC, or ETSI. European Standards are referred to as an “EN standard.” The “EN” is derived from the German name Europäische Norm (i.e., translated as “European Norm”). Each of the member states have their own standards organizations that is responsible for adopting ISO standards that have been ratified, translation of the standard into the language of the member state, and and issue of translated EN standard. For example: German standards are preceded by “DIN,” Irish standards are preceded by “I.S. EN,” and Swedish standards are preceded by “SS-EN.”
How is an EN version different from the ISO version of a standard?
Historically, EN medical device standards include three Annexes related to harmonization with the standard with the three EU Directives (i.e., MDD, AIMD, and IVDD). Now that the EU Device Regulations have been released (i.e., MDR = 2017/745) and (IVDR = 2017/746), the EN standards now have harmonization Annexes for the two regulations (i.e., ZA and ZB). The content of the ISO Standard is usually not changed in an EN standard unless there is a correction, amendment, or deviation.
Where can you purchase EN versions?
EN standards are translated from the adopted ISO standard by the standards organization for each member state. Below are a few examples:
If you already own the ISO version, do you need to buy the EN standard too?
In a Technical File for CE Marking, you are required to demonstrate how you comply with Annex I of the MDR or IVDR. The recommended method of demonstrating compliance is creating a General Safety and Performance Requirements (GSPRs) checklist. In that checklist, you need to identify which applicable standards were used. If an EN standard is available, the GSPR checklist should reference that standard instead of the ISO version. Unfortunately, in order to claim compliance with the EN version, someone needs access to that standard. This could be within your organization or a consultant that assisted in preparing the GSPR checklist to ensure that you are compliant with the EN standard. Generally, we purchase English versions of EN standards from the Estonian Center for Standards and Accreditation, because it is usually the least expensive source. However, if you ask a consultant to do this comparison for you, the best way to perform that comparison is using the comparison function of Adobe Acrobat Pro.
Can you identify EN requirements without the EN version?
Most of the quality system requirements for the MDR and IVDR regulations are found in Article 10 of the regulation. However, there are quality system requirements found in other articles and in the annexes. Therefore, you may be able to find EU requirements in the regulations by doing a keyword search. For example, searching for the word “risk” may help you find risk management requirements throughout the regulations. You may also find European-specific requirements in Common Specifications and MDCG guidance documents.
How to respond to a certification body auditor?
Final Answer: I’m not sure, because every auditor is a little different in their approach. However, as an instructor, I would teach an auditor to ask open-ended questions, such as: “How did you determine if there is an impact upon your procedures and design documentation with regard to the EN standard?” (i.e., – impact analysis). If the company provides an impact analysis and explains why the existing documentation and procedure should not change, I believe this meets the EU requirements. If the certification body auditor is still not satisfied, then you might try asking them, “What differences are you aware of between the EN and ISO versions of the standard?”
Learn how to create a regulatory plan for combining 510k with CE marking submissions in parallel instead of doubling your workload.
My first medical device regulatory submission was for CE Marking, while my second regulatory submission was for a 510k submission of the same product. Preparing submissions for different countries in parallel is a common path for medical device regulatory submissions, but it is also an inefficient path. If you know that you will be submitting both types of documents, then you should plan for this from the start and reduce your workload by at least 35%.
The reason why you can quickly reduce your workload by more than 65% is that both submission have very similar sections. Therefore, you can write the content for those sections in such a way that the material can be used for your 510k submission and CE Marking.
Identify duplicate sections in when combining 510k with CE marking projects
Most of your testing requirements should be identical when you are combining 510k with CE Marking submissions. However, the way the testing is presented is different. For your 510k submission you will attach the full testing report and write a brief statement about how the testing supports substantial equivalence. In contrast, CE marking technical files require a summary technical document or STED. The STED is a summary of each test that was performed. If you aren’t sure what testing is required, we created a test plan webinar to address this question specifically. Most of the work will be duplicated between your two test plans, but any outliers should be identified. For example, biocompatibility will need to include a biological evaluation plan (BEP) and biological evaluation report (BER), but this is optional for a 510k submission. There are also FDA requirements that are not required for CE marking, such as material mediated pyrogenicity testing and bacterial endotoxin testing for each production lot. In general, the possible testing categories are:
How to organize your medical device files when combining 510k with CE marking
The new FDA eSTAR has a unique PDF template that must be used for organizing your submission but Patrick Axtell, the person that helped create the FDA eSTAR templates, is also the Coordinator for the IMDRF Regulated Product Submission Working Group. He has inserted links in the eSTAR sections that cross-reference to the Regulated Product Submission Table of Content (i.e., RPS ToC). Therefore, best practice is to organize your medical device file in accordance with the RPS ToC:
Combining 510k with CE marking – how to construct your regulatory plan
In one of my previous blogs, I explained how the new FDA eSTAR template as a project management tool to verify that all of the section of a 510k submission are complete. Unfortunately, there is no CE Marking equivalent, but you can use your TF/MDR Index as a project management tool when you are constructing a combined plan for a 510k submission and CE Marking. The first step is to create a Index based on a recognized standard (EN standard or ISO standards). Historically we used the GHTF guidance document released by study group 1: N011:2008. When the EU MDR came into force, we added cross-references to the EU MDR in our TF/MDR Index. The GHTF guidance mirrors the format required in Annex III of the new EU MDR. I do not recommend using the NB-MED 2.5.1/rec 5 guidance document. Even though the content is similar to the GHTF guidance, the format is quite different. There is also a new IMDRF guidance document for Essential Principles of Safety and Performance that you should consider referencing.
Project and task management for your combined regulatory plan
If you are going to outsource sections of either submission, the sections should be written and reviewed by someone familiar with both types of submissions. The headers and footers will be unique to the type of submission, but I write the text in Google Docs without formatting for ease of sharing and so I can use my Chromebook.
If you have an in-house team that prepares your 510k submissions and Technical Files, you might consider training the people responsible for each section on the requirements for each type of submission. This eliminates rewriting and reformatting later. I like to assign who is writing each section in a separate column of my project management software. Then I will sort the sections by the expected date of completion. All the safety and performance testing, and any sections requiring validation, will typically be finished at the end of the project. Therefore, it is important to dedicate unique resources to those sections rather than asking one person to write several of those sections. You also will want to make sure any supporting documentation they need is completed early so that the project’s critical path doesn’t change.
Additional training for combining 510k with CE marking
We provide an on-demand 510k course series consisting of 33 FDA eSTAR webinars that you can purchase as a bundle or individually. We also have various training webinars about CE marking on our webinars page.
In this article you will learn tips and best practices for creating and maintaining your DHF (i.e., Design History File).
What is a DHF?
DHF is an acronym for design history file. The US FDA is the only country that specifically includes this in medical device regulations (i.e., 21 CFR 820.30j). Other countries simply require that you maintain records of design and development. The DHF is a file, virtual or physical, that includes all the records of your efforts to design and develop a medical device. You could create an index for a DHF that includes all of the most recent versions of the documents pertaining to the design of your device, but that is called a Device Master Record or DMR (i.e., 21 CFR 820.181). Another term for the DMR is “technical file” or “medical device file.” The FDA will be adopting the term “medical device file” as part of the Quality Management System Regulation (QMSR), and the definition for a DHF will become obsolete February 2, 2026.
What’s the difference between a DHF, DMR, and DHR?
The US FDA loves acronyms, but overuse of acronyms leads to confusion. The acronyms DHF, DMR, and DHR are the three most commonly confused acronyms in the medical device industry. One of the simplest solutions is to stop using acronyms and to rename each of these documents so that they are no so confusing. Instead of design history file, or DHF, start referring to this file as a record of new product development. That record consists of thousands of pages covering a year or more of design and development activity by your new product development team. Instead of device master record, or DMR, start referring to this as your medical device file or technical file. Clause 4.2.3 of ISO 13485:2016 is specific to this record. It is intended to be a living document that you will update each time you make a design change. Instead of device history record, or DHR, start referring to this as your batch record or lot record. This is a file containing all of the records, or cross-references to records associated with the manufacturing and inspection of a batch or lot of devices. The batch record or lot record will be reviewed for completeness, accuracy, and to ensure all inspection and testing passed. Often a release checklist is used to ensure consistency of this review, and the checklist will be signed and dated by the person that releases the batch or lot for distribution. For sterile products, this is done after sterilization and sterile product is quarantined until release.
Regulatory Requirements for a Design History File (DHF)
The requirements for a design history file (DHF) are found in 21 CFR 820.30(j):
“Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and the requirements of this part.”
There is also a definition for a DHF found in 21 CFR 820.3(e):
“Design history file (DHF ) means a compilation of records which describes the design history of a finished device.”
The FDA provided an official interpretation of this requirement in the preamble when the QSR was published in 1996. That discussion of the requirement indicated that the DHF is intended to be a repository of the records required to demonstrate compliance with your design plan and your design control procedures. The discussion also indicates that the same DHF may be used for minor variations of a device such as size differences. Most manufacturers will organize the DHF in a binder and organize the binder chronologically to match a design project plan, however, most do not create a DHF template. Meeting minutes fromeach design meeting are typically included as an appendix to the DHF, while reviewed and approved documents such as the design plan, design inputs, design outputs and records of design reviews typically comprise the bulk of the DHF. Manufacturers also typically will conduct an internal auditor of active DHF binders in order to ensure that design projects are following the approved design plans.
Why you should never use a DHF template
The DHF is is intended to provide evidence of following an approved design plan, but the DHF consists of many records–not just one record. A DHF template could be created to follow a standardized design control process, but most manufacturers write a generic design procedure that allows and encourages the design team to customize the design plan to match the needs of each development project. Therefore, design plans may have different numbers of design reviews and very different testing activities prior to the start of the design transfer process and during design verification and validation.
For a device master record (DMR), I recommend creating a DMR Index using a template that is organized in accordance with an international standard to meet the needs of a DMR and a Technical File. The DMR is a living document that only shows the most current design outputs for a device while a DHF may require repeating various verification and validation testing if the initial design fails to meet acceptance criteria and a design change is required prior to the final design review and approval of commercial release. The need for including this variability eliminates the advantages of a template.
What is the purpose of the DHF?
The purpose of the DHF is to provide objective evidence that the design and development team followed design controls in order to develop a medical device. FDA inspectors review the DHF to make sure that the design process is effective by verifying that known hazards were identified, and appropriate design inputs (i.e., test requirements) were approved. The design specifications should demonstrate that risks were reduced as far as possible by implementing design solutions, protective measures, and by providing warnings and precautions of residual risks to users and patients. Verification of design inputs and validation of user needs comprise the bulk of your DHF in the form of testing protocols and reports. Inspectors will review your design transfer activities to ensure that the output of manufacturing consistently meets your design specifications. Design reviews meeting minutes will be sampled to verify that you included an independent reviewer and the design team completed all activities planned in your design plan. FDA inspectors will verify that your design team has adequate training on design controls and the relates processes. Finally, the FDA will look for justifications and updating of design documentation for the design changes your team made during the design process.
Where should you document design changes?
Product design changes that occur prior to the final design review and approval of commercial release are required for inclusion in the DHF. However, once a product is released the control over design changes should be tighter and regulatory submission of changes may be required. Therefore, I recommend documenting post-market design changes in the DMR Index for a device as part of the revision history. I treat the DMR Index as a controlled document and any post-market design changes are reflected in the revision history with a reference to the design change approval (e.g., ECN 123 – addition of UDI label to product labeling). The other advantage of this approach is that all post-market design changes that must be documented for a design dossier are summarized in the revision history of the DMR Index and the DMR Index will serve as a Technical File/Design Dossier.
Training Webinar
If you are interested in learning more about design history files, we recorded a DHF training webinar. The webinar explains how and when to create a design history file (DHF). After you create a design control procedure, you can show the recording of this webinar to your design and development team to ensure that design and development documentation is compliant and updates are efficiently maintained. We are in the process of updating this webinar and it will be hosted live in September.
This article explains how to write your classification recommendation for a De Novo Classification Request using a risk-based approach.
“Automatic Class III Designation” does not mean that your device is a Class III device. That phrase means that the device is new, and therefore it will be automatically classified as Class III until a company submits a De Novo Classification Request. You and your company, not the FDA, should make the classification recommendation and propose the regulatory pathway for a new device. Submitting a 513g request is an option, but a 513g request involves paying the FDA money to write a classification recommendation. The FDA will always be more conservative in their assessment than the manufacturer.
Although no FDA guidance explains how to write a classification recommendation, companies have been writing these documents for years–for Technical Files. Most countries have risk-based classification rules, while the FDA’s product classification database is centered upon precedents and adjusted over time by historical trends of adverse events and recalls. Therefore, you should write a classification recommendation for the FDA that is focused on a documented risk assessment. Your approach will also need to be modified to include classification information for similar indications for use and technological characteristics that are already established in the US market.
Most Common Mistakes in Writing a Classification Rationale
Many people mistakenly write a short classification rationale for a technical file, which simply states which classification rule applies and why. Although this approach is acceptable for a Declaration of Conformity, you must provide a comprehensive classification rationale in your technical file. First, you need to make sure that there is only one classification rule that applies. For example, classification rules fall into four general categories:
Non-invasive Devices
Invasive Devices
Active Devices
Special Rules
The software was haphazardly added to the active devices category until recently, and special rules were created to address emerging areas of interest and concern. Therefore, most active devices have a second rule that applies regarding the invasive nature of the device–or lack thereof. In order to write a comprehensive classification rationale, you need to review each classification rule and document your explanation for why it applies or does not apply to your device.
A Classification Recommendation Compares Indications for Use
The FDA does have classification rules, but the rules are not 13 numbered items in the Code of Federal Regulations (CFR). The FDA expects a risk assessment of comparing your device with existing devices on the US market. The basis of comparison should be: 1) the indications for use and 2) the technological characteristics. First, you should identify other devices that have similar indications for use. For example, a device intended for home use or over-the-counter (OTC) use represents a higher risk to patients and users than a device intended for prescription use only. Patients may fail to identify contraindications for a device properly, or the lack of formal medical training may result in use errors that would not occur when a physician uses the same device.
Other aspects of indications for use that impact the risk assessment are the part of the body where your device will be used and the duration of use. For example, implants are at higher risk than non-implants, because implants are in contact with the body for a much longer period of time. Implants can also expose the body to systemic risks, while a surface contacting device is likely only to have a localized effect. Degradation of implants also exposes the body to small particles, with more surface area, that can travel from one part of the body to another.
If your device is used for life support, the device will also be considered at higher risk than devices that are not required for life support. If your device is the only device used for diagnosis, this also represents a higher risk than a device that acts as an adjunct to other devices. Finally, if your device is an accessory to other devices that are high risk, your device may be considered a higher risk as well–especially if it controls the higher risk device.
In your analysis, you need to identify devices that are already on the US market that have similar indications for use. Usually, those devices will be Class II devices. However, if some of those devices are Class I or Class III, you will need to be more careful with how you differentiate your indications for use from those other devices.
A Classification Recommendation Compares Technological Characteristics
When comparing technological characteristics, the following aspects should be considered: 1) materials, 2) design, 3) energy source, and 4) other design features. For example, absorbable materials are generally considered at higher risk than devices that are not absorbable. Sterile devices are generally at higher risk than non-sterile devices because the failure of the sterilization process or the package integrity can result in serious infections and death. Devices that are electrically powered are usually considered at higher risk than devices that are not powered. Finally, software-controlled devices that provide feedback control are considered at higher risk than a device that does not have feedback control. Each technological characteristic also represents a different category of hazard. Hazard categories are listed in Table C1 of Annex C in ISO 14971:2019. These include chemical, biological, electrical, radiation, etc.
Once you have identified the Classification of other devices with similar indications for use and technological characteristics, you need to estimate the risks for each hazard identified. This involves more than just listing hazards and assigning scores for severity and probability for the occurrence of harm. Severity should consider the type of injuries, the number of injuries, and the duration of harm. Probability should consider the frequency of events (P1), and the probability of events resulting in injury (P2). These risk estimates also require clinical data.
Benefit-Risk Analysis
In the end, you prepare a benefit-risk analysis for your device. This is much more than a statement that the benefits outweigh the risks. You need to identify the clinical benefits of your device when compared to alternative treatments. You also need to analyze risks relative to alternative treatments. You will need to prepare this as a summary of risks–not a list of hazards. Ultimately, your benefits should be equivalent to the benefits of existing devices on the market or better, and the risks should be equivalent to existing devices on the market or less.
Examples of Classification Recommendation
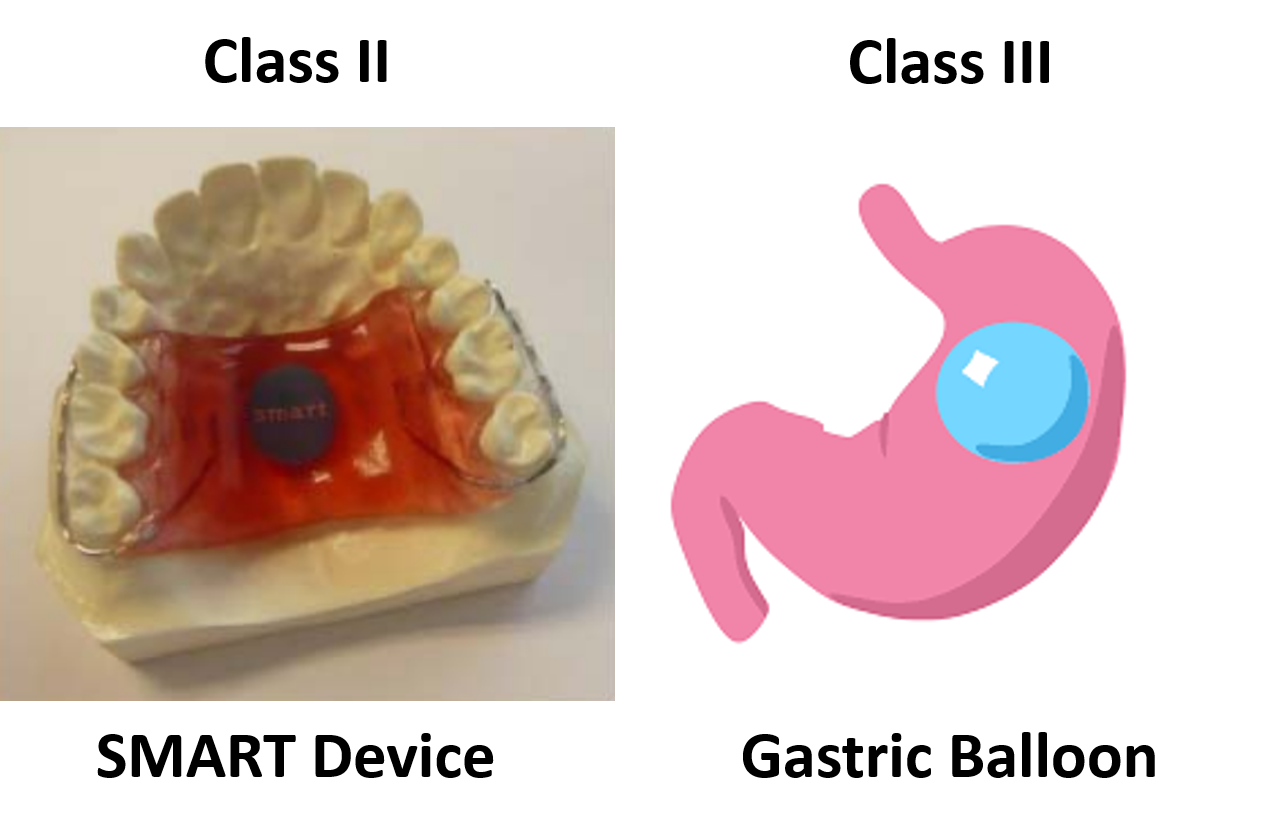
Eight different medical devices are legally marketed in the USA for weight loss or weight management:
The indications for use for these products are similar, but not identical. Plenity is indicated for patients with a BMI of 25 – 40 kg/m2. In comparison, ORBERA is indicated for patients with a BMI of 30-40 kg/m2, and AspireAssist is indicated for patients with a BMI of 35-55 kg/m2. All three of these indications have overlapping BMI ranges. However, the clinical benefits to a person with a BMI of 25 kg/m2 are not the same as the clinical benefits to a person with a BMI of 40 or 50 kg/m2. Therefore, these minor differences in BMI can have a significant impact on the benefit/risk analysis used for a De Novo approval decision and the Classification (i.e., Class II or Class III) determined by the FDA.
The only two weight management devices that received the approval of the De Novo Classification Request had very different technological characteristics from the other six devices. All six Class III, PMA devices, are implants, while the Class II devices are not implants. The risks associated with implants are much greater than with non-implants. The risk of implants breaking or leaking, and the difficulty in removing an implant, are just two of the considerations that must be evaluated in deciding whether an implantable device should be a Class II or Class III device.
This article reviews the top reasons why other companies feel requesting FDA pre-sub meetings is a waste of time but you can’t afford to.
It only takes our consulting team a few hours to prepare an FDA pre-sub request using the new FDA PreSTAR. The FDA does not charge you a cent for submitting an FDA pre-sub, and a pre-sub should be part of every design plan. However, most companies are resistant to requesting FDA pre-sub meetings. In this article we review the top four reasons why companies resist submitting an FDA pre-sub request, and you will learn about the three options for the method of FDA feedback.
Our design is not finalized yet
The most common reason why people delay their request for an FDA pre-sub is that they are waiting until the design is complete. This rationale is flawed because the FDA can’t review data and the FDA can’t give you advice on the design of your device. The purpose of an FDA pre-sub is to “forge a better test plan.” The FDA prefers that you submit draft test protocols with specific questions. The ideal time to submit an FDA pre-sub request is 6-9 months before you approve your design outputs (i.e., design freeze). This timing should be shortly after you approve your design inputs (i.e., essential requirements for safety and performance, standards, and stakeholder requirements). Another reason for requesting a pre-sub early is that most companies need to submit a pre-sub supplement with a revised test plan. This is especially true for biocompatibility testing, non-clinical, benchtop performance testing plans, pre-clinical animal testing, human clinical studies, and human factors testing. The revised test plan includes protocol revisions based on the feedback from the original pre-sub.
It’s too late to request FDA pre-sub meetings
If you are less than a week away from submitting to the FDA, it is too late. The FDA target for scheduling an FDA pre-sub meeting is 70-75 days from the date your request was submitted. That’s 10-11 weeks. Most companies tell me that they plan to submit to the FDA within weeks or a couple of months, but most of the companies take nine months or longer. For example, what if your device fails EMC testing, and you have to change the design and retest for both EMC and electrical safety at an NRTL? At best, you will have an 8-week delay. If you submit a request next week, and everything goes as planned, you can always withdraw your request for the pre-sub. If you encounter a delay for any reason, suddenly, it’s not too late.
We don’t want to be bound by what the FDA says in the FDA pre-sub meeting
FDA pre-sub meetings are “non-binding.” That means that the FDA can change its mind, but it also means you don’t have to do everything the FDA says in an FDA pre-sub meeting. If you don’t ask a question about testing requirements, that doesn’t mean that the FDA does not have any testing requirements. The FDA knows what previous companies have submitted for testing better than you do, and they may be in the process of evaluating draft guidance documents. If you ask questions, you will have better insights into what the FDA expects. The most important questions are related to your rationale for why a specific testing specimen represents the “worst-case” for one of your tests. Selecting the wrong testing specimen will result in you repeating that test. Understanding FDA expectations helps you write better rationales for testing or test avoidance. You also might learn about deadlines for the implementation of new testing requirements that you might be able to avoid. Finally, you can ask the FDA about possible testing options you are considering if the FDA denies your most optimistic testing plans.
There is already a guidance document for our device
Not all device classifications have a guidance document explaining what information should be submitted in an FDA pre-market submission. However, there are almost one hundred Special Controls Guidance Documents, and for new device regulations (i.e., De Novo applications) the FDA now incorporates the special controls directly in the new regulation. Therefore, there is a good chance that the FDA published special controls as part of the regulation for your device or as a guidance document. As part of the special controls, the FDA defines what performance testing is required for your device. If you already know what testing is required, then the value in requesting FDA pre-sub meetings is diminished. But at least three other key benefits remain.
First, you can verify that the predicate you plan to use for comparative testing is not going to be a problem. Although the FDA can’t tell you which predicate to pick, the FDA can tell you if there is a problem with the predicate you have selected. This is especially important if the product is not currently registered and listed, because you may not know if the device was withdrawn from the market after it was cleared.
Second, not all testing standards are prescriptive. Many tests have testing options that require a decision. Input from the FDA may be valuable in making choices between various performance testing options. Sometimes, you forgo testing and provide a rationale instead. FDA feedback on any rationale for not doing testing is critical to prevent delays and requests for additional information later.
Third, there are many different FDA representatives who participate in FDA pre-sub meetings. The lead reviewer will invite specialists and the branch chief to the meeting. Each of these specialists can answer questions during a pre-submission meeting that they are not able to answer during the actual review process. You also have the opportunity to get feedback from the branch chief–who has insight from all the previous devices that were cleared with your product classification. Your lead reviewer is not likely to be as experienced as the branch chief, and may only have been working at the FDA for months. Your request for the 510k pre-sub meeting will help an inexperienced lead reviewer as much as it will help your company.
Which method of FDA pre-sub feedback should you request?
The FDA offers three different options for the method of feedback in pre-sub request:
a face-to-face meeting
a conference call
an email response
Feedback Option 1 – A Face-to-Face Meeting
Some executives believe that face-to-face meetings are critical in establishing relationships with people. However, you need to understand the culture of the people you are trying to build a relationship with. The FDA is an overworked bureaucracy, and government agencies have security concerns. When the FDA meets with visitors they must go to a different building and arrange for their guests to pass through security. This is more work and takes more time. To justify the extra work and time, you need a compelling reason why a face-to-face meeting with the FDA is necessary.
Traveling to the FDA will cost your team money and time that conference calls and emails will not. More importantly, you are limited to one hour for a pre-submission meeting. One hour is barely enough time to ask questions and listen to the answers. You only have minutes to introduce your company and your team and describe the product. There is no time for relationship building. The best way to impress the FDA is to 1) prepare thoroughly, 2) conduct an efficient meeting, and 3) ask smart questions.
There is one time when you should visit the FDA face-to-face–if you have a powerful demonstration and video just isn’t good enough.
Feedback Option 2 – Conference Call
Conference calls save you time and money, but conference calls also save the FDA time and effort. You won’t personally meet people from the agency, but you can communicate information prior to the meeting and you can provide videos of simulated use for your device. Conference calls do have the advantage of allowing you to mute the call for a moment and make a comment among your team members without the agency listening as well. Whenever you are discussing a performance testing plan or a clinical study protocol with the FDA, you will probably want a conference call to enable clarification questions. The image below is an example of a request for a teleconference as the method of feedback. The FDA will still send an email response to your questions within 70 days, and you will want to submit your presentation slide deck for the teleconference at least 48 hours before the teleconference.
Feedback option 3 – Email
Email responses from the FDA are highly underrated in value. When you specify an email response, you generally receive a response to your questions sooner. You also should receive more information, because each person from the agency is able to provide an hour of their time to write detailed feedback. In a conference call, you are speaking for part of the hour, and only one person from the FDA can speak at a time. Therefore, you almost always have less feedback during conference calls and face-to-face meetings. The primary downside to email as a feedback method is that it is not interactive. In the case of submission issue review (SIR) requests (i.e., a special type of FDA pre-sub request), the FDA will only give you the option of a teleconference or email feedback–not both. Due to the challenges of scheduling teleconference, sometimes an email response can be delivered sooner and might be your best choice for an SIR request.
Learning More about FDA Pre-sub Meetings
If you would like to learn more about how to prepare the format and contents of an FDA pre-sub request, we have a four-part webinar series that will teach you how to prepare a pre-submission meeting request using the new FDA PreSTAR template.