The FDA Breakthrough Device Designation was created in 2015 to expedite device access for life-threatening and debilitating diseases.
What is the FDA Breakthrough Device Designation?
The FDA Breakthrough Device Designation is a formal identification by the US FDA that a device in development should be expedited for patient access because it has a reasonable chance of providing more effective treatment than the standard of care for the treatment or diagnosis of life-threatening or irreversibly debilitating human disease or conditions.

To be granted breakthrough status, your device must also meet at least one of the following four secondary criteria:
- Represents Breakthrough Technology
- No Approved or Cleared Alternatives Exist
- Offers Significant Advantages over Existing Approved or Cleared Alternatives
- Device Availability is in the Best Interest of Patients
Once the FDA has designated your device as a breakthrough device, all future communications with the FDA related to that device should be identified with the Q-sub reference number assigned to your breakthrough request. If you want more information, please schedule a call with us, or you can download the FDA guidance. We have helped multiple clients successfully receive breakthrough device designation.
What are the benefits of receiving the designation?
The breakthrough designation helps the FDA identify new technology to focus on to expedite access to novel devices that will save lives and treat debilitating diseases. It takes the FDA longer to review these devices because they may raise novel scientific and regulatory issues. Therefore, the FDA prioritizes 510k and De Novo submissions for breakthrough devices over other 510k and De Novo submissions, and the FDA’s senior management is involved in the review process. The average review time for the 32 breakthrough devices with 510k clearance was 152 days*. This may not seem like an expedited review, but the average review time for 510k cleared devices that require additional testing data is almost 270 days. The average review time for the twenty De Novo Classification Requests designated as breakthrough devices was 312 days*. This significantly improved compared to the average De Novo Decision timeline of 390 days for 2019-2023.
*Metrics updated on 10/31/2022 with data through 9/30/2022
Are there reimbursement benefits?
There have been multiple proposals to offer earlier reimbursement for Breakthrough Device Designation. Typically, CMS does not cover new technology for the first two years. Specifically, the Centers for Medicare and Medicaid Services (CMS) typically takes two years to establish qualification for public reimbursement coverage in the USA. In contrast, private insurers are inconsistent in their coverage because Medicare Administrative Contractor (MAC) is divided into 13 different US regions, each making independent coverage decisions case-by-case. Unfortunately, none of the proposed bills for immediate coverage through CMS have been approved.
Mechanisms of Expedited FDA Review
In addition to identifying breakthrough devices for priority review and involving the FDA’s senior management, the FDA also offers four other mechanisms for improving the review time. First, the FDA offers “Sprint discussions.” A “Sprint” discussion allows the FDA and the company to discuss a single topic and reach an agreement in a set period (e.g., 45 days). The FDA provides an example of a Sprint discussion, such as a pre-submission meeting. Still, the timeline is half the duration of the FDA’s target MDUFA V decision goals.
The second mechanism for improving the review time is a Data Development Plan (DDP). Using this mechanism, the FDA will work with the company to finalize the breakthrough device’s non-clinical and clinical testing plans. This may include starting clinical testing earlier while deferring certain non-clinical testing.
The third mechanism for improving the review time is the Clinical Protocol Agreement. In this scenario, the FDA will interactively review changes to clinical protocols rather than conducting a protocol acceptance review first. Therefore, the time required to review and approve a clinical protocol change is less, and the sponsor can complete their clinical studies in less time.
The fourth mechanism for improving the review time is a prioritized pre-submission review. If a company prefers to discuss multiple issues in one meeting rather than conducting Sprint discussions on single topics, then the FDA will prioritize pre-submission review. The prioritized pre-submission will be tracked as an interactive review with a shorter timeline than other pre-submission meeting requests.
How do you apply to the FDA for Breakthrough Device Designation?
To receive the designation, you must prepare a Breakthrough Device Designation request and submit it to the FDA Document Control Center (DCC) as an eCopy. The eCopy can be done via FedEx or through the new Customer Collaboration Portal (CCP) launched by the FDA in 2022. Your application could consist of a single document, but we recommend at least three documents: 1) a formal request outlining how your device meets the criteria for breakthrough designation, 2) a detailed device description, and 3) preliminary clinical data demonstrating the feasibility of your device delivering performance claimed in your request for designation. There are no user fees associated with the application for breakthrough designation, and you are not prevented from submitting other types of submissions in parallel with the breakthrough designation request, such as a pre-submission or investigational device exemption (IDE).
When should you apply to the FDA?
If the FDA denies an initial breakthrough designation request, the company may re-submit a request later. Therefore, companies should submit requests as soon as they can provide preliminary clinical data to demonstrate the feasibility of the device’s claimed performance. Therefore, a breakthrough designation request would typically be submitted after an Early Feasibility Study (EFS), which allows a maximum of ten clinical subjects.
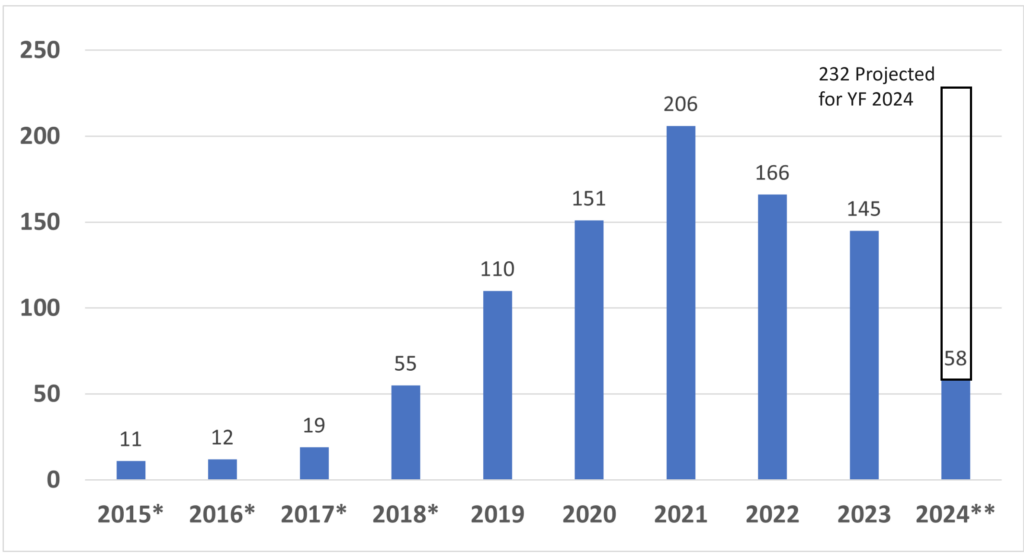
How many companies have received Breakthrough Device Designation from the FDA?
Since starting the Breakthrough Designation program in 2015, the FDA has granted 933 devices Breakthrough Device Designation*. CDRH, the device division of the FDA, granted 921, while CBER, the biologics division of the FDA, granted 12*. The breakthrough device designation, however, does not guarantee FDA market authorization. Only 95 of the breakthrough designations have resulted in market authorization so far. Four of the 95 devices were reviewed by CBER. Of the remaining 91 devices, 32 received 510k clearance, 30 De Novo Classification Requests were granted, and 31 PMAs were approved*. Given the number of submissions received yearly, only 10-15% of De Novo and PMA submissions are also Breakthrough Devices. In contrast, only about 0.1% of 510k submissions are also Breakthrough Devices. The data for breakthrough device designation is only reported through December 31, 2023, but the projected number of breakthrough designations for FY 2024 (ending September 30, 2024) is 232.
*Metrics updated on 4/14/2024 with data through 12/31/2024
**FY 2024 data is limited to one quarter
Pingback: FDA on pace for a Record Number of Breakthrough Devices in 2023 – BiomedSpecialist
Thanks for the video overview on Breakthrough – your company sounds very interesting, and I enjoyed the self-disclosure that you are “irreverent” – because I am as well. I have a boutique reimbursement consulting business and have several start up clients, and one of them pointed me to your content. I just wanted to point out that your reimbursement section is slightly misleading. Breakthrough status doesn’t provide immediate coverage once you have FDA clearance – it might in the future, but MCIT was discarded by CMS, and the TCET is still in rule making, so there could be a time when that’s true, but it isn’t now. Also, you might want to mention that the hospital inpatient New Tech Add On Payment (NTAP) and outpatient Transitional Pass Through payments are made easier if a product has Breakthrough designation.
Just wanted you to be aware, and feel free to email me if you need any back up documentation!
Thank you for the great comment Tina. Yes, when we created the content it appeared that there were be additional reimbursement benefits to obtaining Breakthrough Designation, but that has not been approved.
It seems the FDA’s eligibility criteria are very broad, but then in practice, they pick and choose.
The FDA has a different set of time data, “The average review time for the seventeen 510k cleared breakthrough devices was 155 days*. This may not seem like an expedited review, but the average review time for 510k cleared devices that require additional testing data is almost 270 days. https://medicaldeviceacademy.com/breakthrough-device-designation” Note the include the caveat, “for 510k cleared devices that require additional testing data”
The reason for longer review times is almost always additional testing data is needed. For devices that require clinical data this is very common (not pick and choose). This is why the IVD submissions are longer in general than non-IVD submissions. It is also why Breakthrough devices are longer. We will also be hosting a new webinar on Breakthrough devices in the near future.