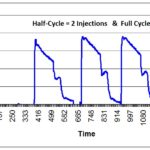
How to write a request for designation
In the eSTAR and PreSTAR, the FDA inquires whether a request for designation (RFD) is associated with your device.
What is a request for designation (RFD)?
A request for designation is a formal request to the Office of Combination Products (OCP), where you request that OCP assign the agency division that will have jurisdiction over a combination product. In 21 CFR 3.7, the FDA outlines the information required in an RFD submission. The FDA encourages RFD submitters to review the agency’s guidance prior to submitting an RFD. It irritates the FDA when you don’t read the guidance and ask questions that are clearly answered within it. Read the guidance first (we provided links below).

What is a combination product?
Before you submit a request for designation, you need to understand what a combination product is. The term combination product includes:
- A product comprised of two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity;
- Two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products;
- A drug, device, or biological product packaged separately that, according to its investigational plan or proposed labeling, is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of the approved product would need to be changed, e.g., to reflect a change in the intended use, dosage form, strength, route of administration, 9or significant change in dose; or
- Any investigational drug, device, or biological product packaged separately that, according to its proposed labeling, is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
Information regarding the drug/biologic constituent part of the combination product may be needed and accounted for throughout the various sections of your premarket submission. In addition, as described in Product Stability documentation, medicinal substance refers to the drug/biologic constituent part of the combination product as defined in 21 CFR 3.2(e).
How do you write a request for designation (RFD)?
We recommend that you always start with a pre-request for designation (pre-RFD). Once you have feedback from the FDA, then you will be ready to write your request for designation (RFD). The FDA published two guidance documents related to RFDs:
What is a pre-RFD?
A pre-RFD is a submission that you make to the Office of Combination products (OCP) to request the FDA’s preliminary, nonbinding assessment of:
- the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and/or
- whether CBER, CDER, or CDRH will regulate the product if it is a non-combination product, or
- which of those Agency Centers will have primary jurisdiction for a premarket submission of a combination product.
The FDA’s target review time is 60 days for providing the information requested, but a pre-RFD is not a tracked metric with budget impact. Therefore, you should set expectations with your senior management team and investors at approximately 90 days–just like the 513(g) submissions.
21 CFR §3.7 – Request for Designation (copied from eCFR)
(a) Who should file: the sponsor of:
- Any combination product the sponsor believes is not covered by an intercenter agreement; or
- Any product where the agency component with primary jurisdiction is unclear or in dispute.
(b) When to file: a sponsor should file a request for designation before filing any application for premarket review, whether an application for marketing approval or a required investigational notice. Sponsors are encouraged to file a request for designation as soon as there is sufficient information for the agency to make a determination.
(c) What to file: an original and two copies of the request for designation must be filed. The request for designation must not exceed 15 pages, including attachments, and must set forth:
- The identity of the sponsor, including company name and address, establishment registration number, company contact person and telephone number.
- A description of the product, including:
- Classification, name of the product and all component products, if applicable;
- Common, generic, or usual name of the product and all component products;
- Proprietary name of the product;
- Identification of any component of the product that already has received premarket approval, is marketed as not being subject to premarket approval, or has received an investigational exemption, the identity of the sponsors, and the status of any discussions or agreements between the sponsors regarding the use of this product as a component of a new combination product.
- Chemical, physical, or biological composition;
- Status and brief reports of the results of developmental work, including animal testing;
- Description of the manufacturing processes, including the sources of all components;
- Proposed use or indications;
- Description of all known modes of action, the sponsor’s identification of the single mode of action that provides the most important therapeutic action of the product, and the basis for that determination.
- Schedule and duration of use;
- Dose and route of administration of drug or biologic;
- Description of related products, including the regulatory status of those related products; and
- Any other relevant information.
- The sponsor’s recommendation as to which agency component should have primary jurisdiction based on the mode of action that provides the most important therapeutic action of the combination product. If the sponsor cannot determine with reasonable certainty which mode of action provides the most important therapeutic action of the combination product, the sponsor’s recommendation must be based on the assignment algorithm set forth in § 3.4(b) and an assessment of the assignment of other combination products the sponsor wishes FDA to consider during the assignment of its combination product.
(d) Where to file: all communications pursuant to this subpart shall be addressed to the attention of the product jurisdiction officer. Such a request, in its mailing cover should be plainly marked “Request for Designation.” Concurrent submissions of electronic copies of Requests for Designation may be addressed to combination@fda.gov.
[56 FR 58756, Nov. 21, 1991, as amended at 68 FR 37077, June 23, 2003; 70 FR 49861, Aug. 25, 2005]
How to write a request for designation Read More »

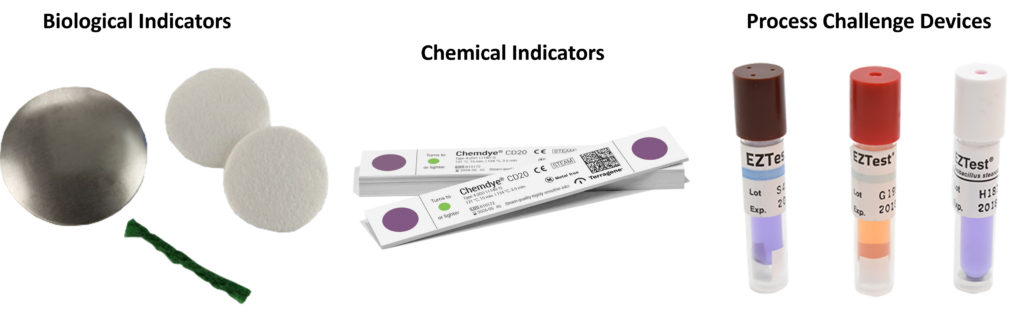
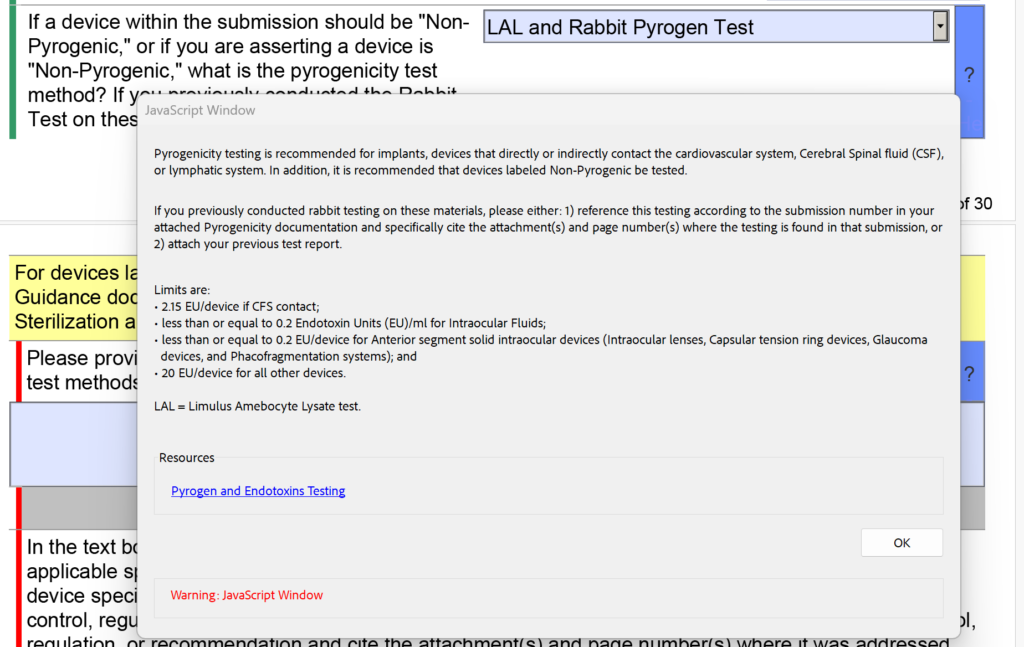
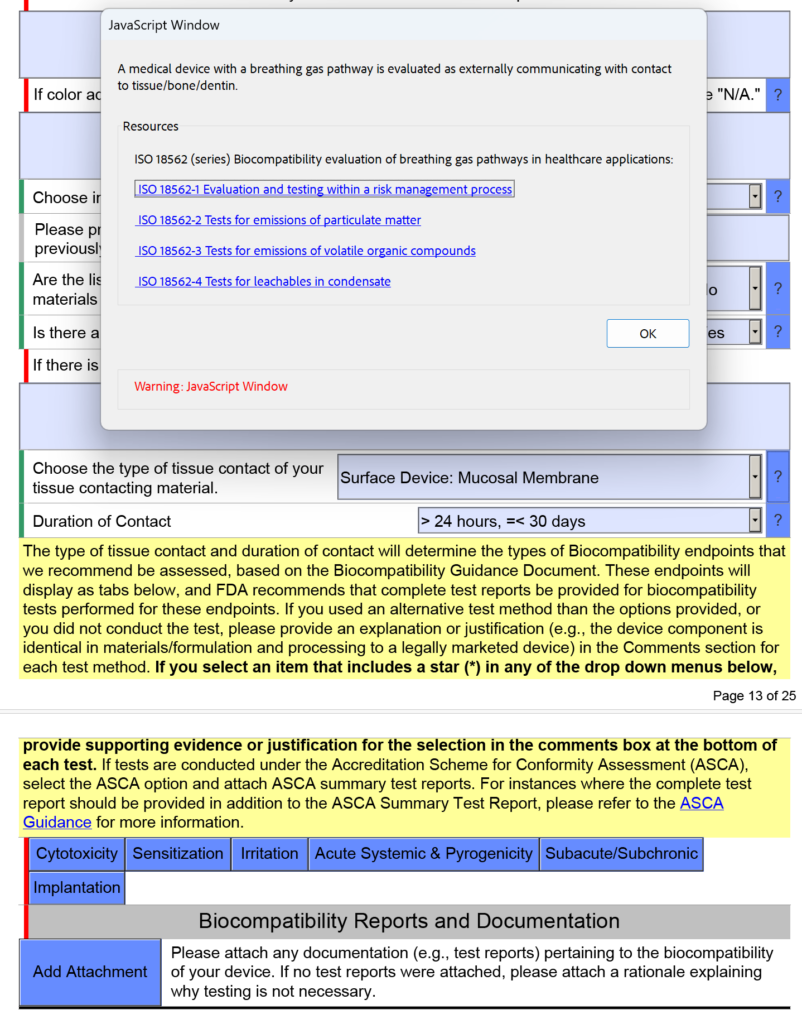
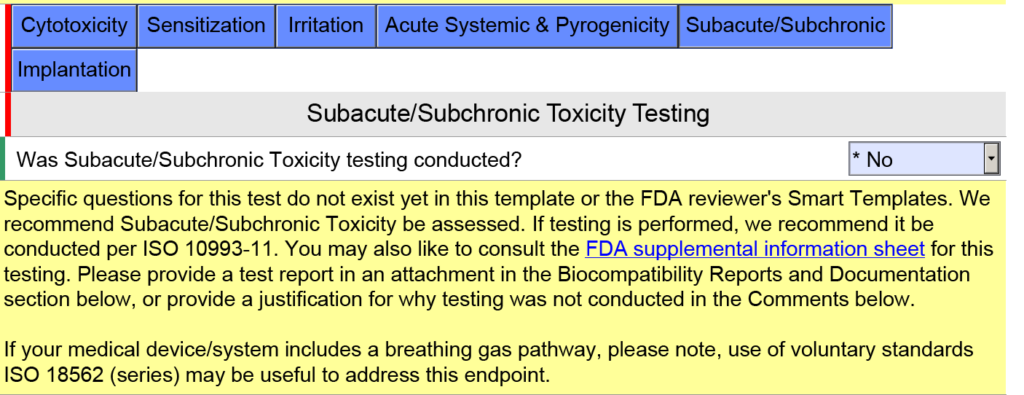
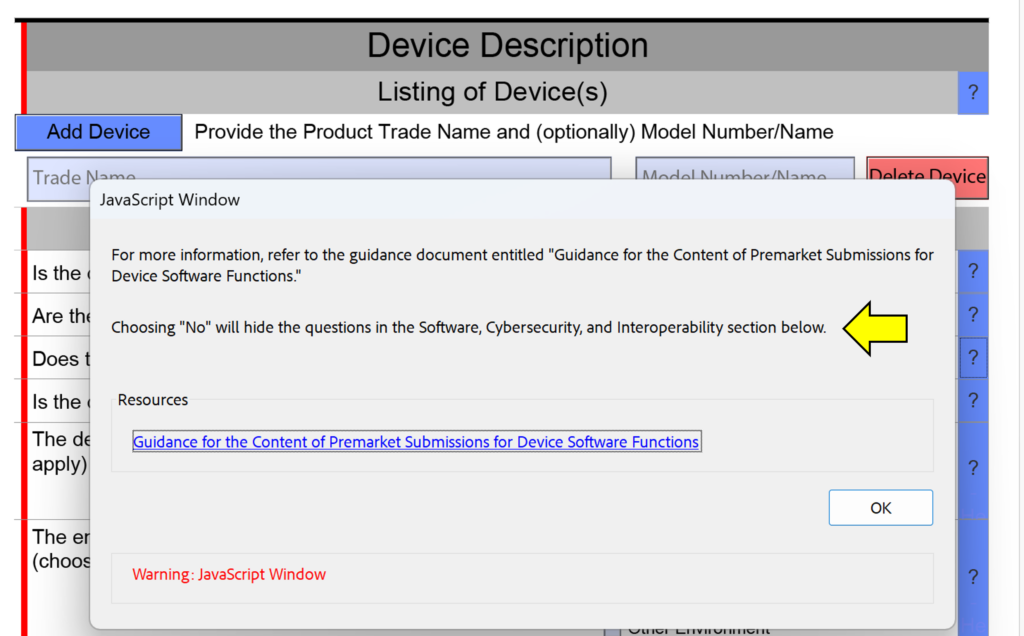
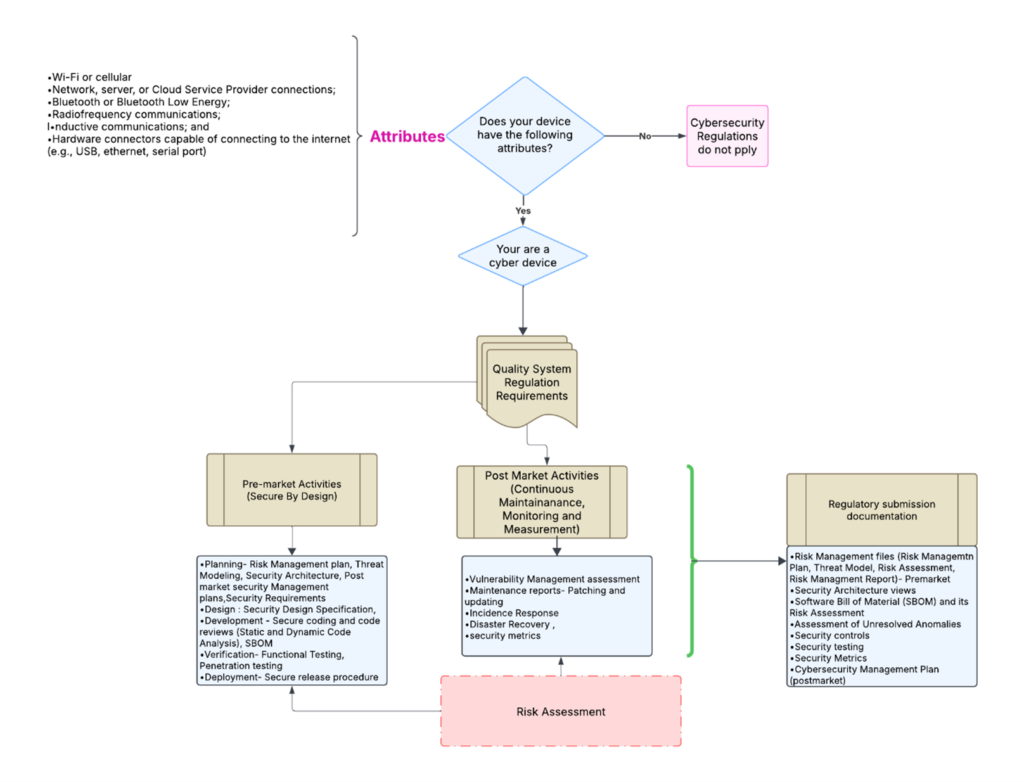
 template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device:
template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device: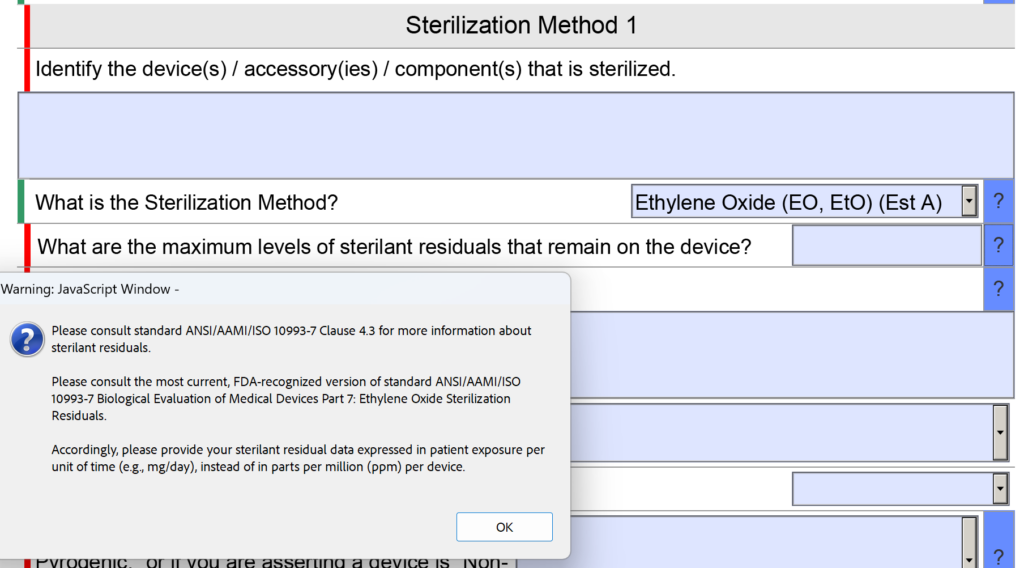




 As the Marketing Lead at BW&CO, Alexina holds a Bachelor of Business Administration in Marketing from the University of Houston. With a deep understanding of the grant funding landscape, she drives strategic marketing initiatives that elevate BW&CO’s brand and reach. Fluent in both English and French, Alexina excels at creating and executing campaigns that connect with diverse audiences. Her expertise includes developing content strategies, managing digital marketing efforts, and conducting market research to support business growth.
As the Marketing Lead at BW&CO, Alexina holds a Bachelor of Business Administration in Marketing from the University of Houston. With a deep understanding of the grant funding landscape, she drives strategic marketing initiatives that elevate BW&CO’s brand and reach. Fluent in both English and French, Alexina excels at creating and executing campaigns that connect with diverse audiences. Her expertise includes developing content strategies, managing digital marketing efforts, and conducting market research to support business growth.