This article discusses the need to requalify EO sterilization validation and explains what is included in our EO sterilization procedure.

Do you need an EO sterilization procedure?
ISO 11135-1 is the international standard for sterilization validation for Ethylene Oxide (EO or EtO) sterilizers. The standard describes multiple methods of sterilization validation: 1) overkill approach, 2) single lot release, and 3) parametric release. The overkill approach is the most common method for validation of your EO sterilization process. The overkill approach is the method recommended by Medical Device Academy’s EO sterilization procedure. If you use a contract sterilizer, the sterilizer will already have completed an Installation Qualification (IQ) and an Operational Qualification (OQ). You must complete a Performance Qualification (PQ) for your product. A typical PQ for initial process validation consists of the following:
- Process Challenge Device (PCD) validation
- Bioburden measurement
- EO residual measurement (as per ISO 10993-7)
- Fractional Cycle (at least one)
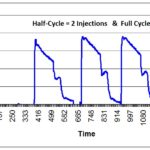
- 3 Half Cycles
- 3 Full Cycles (or 1 Full Cycle, if performed in parallel with the three half-cycles)
Purchase the EO Sterilization Validation Procedure (SYS-031) – $299
SYS-031 EO Sterilization Validation Procedure
This procedure was updated in 2024 to include recent versions of various standards and to incorporate changes to make the procedure consistent with other procedures in Medical Device Academy's turnkey quality system. The updated procedure defines the requirements for ethylene oxide (EO) sterilization validation and revalidation/requalification outsourced to a contract sterilizer.
Price: $299.00
What do MPQ and PPQ mean?
In the ISO 11135 standard, steps #4 and #5 listed above are referred to as the microbial performance qualification (MPQ), while #6 is the physical performance qualification (PPQ). For a successful MPQ, some PCDs must be non-sterile after a fractional cycle to demonstrate the ability to recover the BI challenge organism. After a half-cycle, however, all biological indicators should be sterile.

What are BIs, CIs, and PCDs?
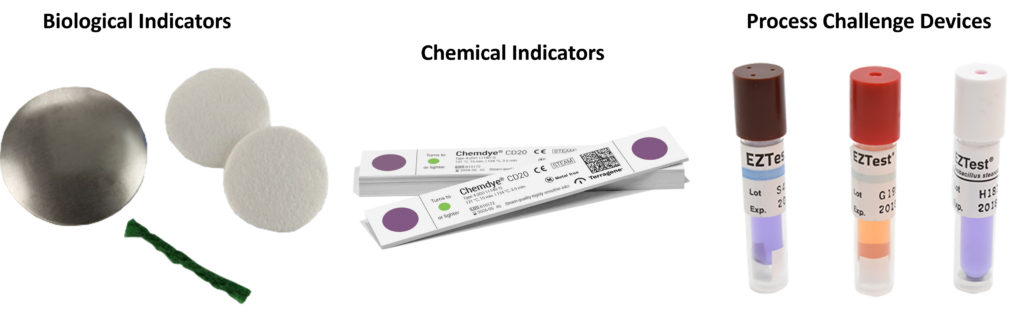
To avoid destructive testing, EO sterilization processes verify sterility by using process challenge devices (PCDs) located outside the device’s primary and secondary packaging. PCDs are more challenging to sterilize than the native bioburden on your device, and the PCDs can be quickly removed from a sterilized pallet without disturbing the wrapping of the pallet. PCDs are also referred to as external biological indicators (BIs). Biological indicators are used internally and externally to your primary sterile barrier packaging during the EO sterilization validation process, but only external BIs are used during routine EO sterilization. You can create your own customized PCD for devices that are especially difficult to EO sterilize (e.g., stopcocks), but you must verify that the PCDs are more challenging to kill than an internal BI in a fractional cycle. Commercially available PCDs often incorporate a chemical indicator (CI) into the label or the cap of the PCD, and some are incorporated into sophisticated tracking software with automatic incubators that read a barcode on the PCD label and detect the results of incubating the BIs in media. These systems are rapid, self-contained BIs that provide validated results in hours, minutes, or seconds.
Outsourcing EO sterilization and requalification
Ethylene oxide sterilization is usually outsourced to a contract sterilizer due to the environmental and safety requirements of working with EO. The contract sterilizer will provide a generic protocol for full validation that is compliant with ISO 11135-1. However, the ISO 11135-1 standard requires that manufacturers perform annual process reviews to evaluate the need to requalify/re-validate the sterilization process. Assuming there have been no problems or changes to the product or EO sterilization process, re-validation is not required at the end of the first year. However, companies are required to re-validate the process after two years–even if there have been no changes.
Longer frequencies for requalification cycles
If there have been no changes to the sterilization process, the product, or the biological indicators, then the manufacturer can use this as a justification for waiting until two years have elapsed before re-validating the ethylene oxide sterilization process. Also, there should be no evidence of sterilization failures or other problems with the validated process. However, that alone is not necessarily enough to justify extending the duration between validations beyond two years. Companies that justify intervals of three or more years have multiple products that use the same EO sterilization process.
In this case, the manufacturer may alternate annually between three, four, or even five different product families that are using the same EO sterilization process. In this case, one of the product families is being re-validated each year or every two years, but the interval between validations for any one product family is longer. This approach is valid if the products are made of similar materials and use the same EO sterilization process. If you only have one product, then you need to re-validate the sterilization process once every two years to verify the process remains active.
Minimum revalidation requirements
When you determine that it is time to re-validate your ethylene oxide sterilization process, you need to perform the following tests to meet the minimum requirements of ISO 11135-1:
- Re-validation of PCD
- Bioburden measurement
- EO residual measurement
- 1 Half Cycle
- 1 Full cycle (to verify the EO residuals are acceptable)
The purpose of #1 is to verify that the resistance of internal BIs used in the half-cycle is more resistant than the product bioburden. The purpose of #2 is to verify that bioburden levels have not changed, and the type of organisms has not changed. In practice, most companies monitor bioburden quarterly, and therefore this step should be routine. Step 3, EO residual measurement, should be performed to verify that there have not been minor changes to the product or process that would increase the concentration of EO, Ethylene Chlorohydrin (ECH), or Ethylene Glycol (EG) beyond the Tolerable Contact Limit (TCL). The purpose of this third test is to prevent localized irritation caused by residual chemicals from the ethylene oxide sterilization process.
Step 4 of the re-validation is intended to verify that a full injection of EO is more than required to kill the bioburden present for the number of injections required for a half-cycle.
The final step is to perform a full cycle. The product from the full cycle is typically used for EO residual testing. Any product from the full cycle that is not used for testing can be sold after sterility testing is complete.
Partial loads & rework
If you occasionally sterilize loads that are less than “full loads,” then you need to ensure that you have validated a minimum load or a specific partial load (e.g., half-pallet, instead of a full pallet). In the case of a partial or minimum load, you may identify different locations in your load that is considered “worst-case.” These are the locations that had PCDs that were not sterile in a fractional cycle.
Most companies do not have concerns about the cost of the actual sterilization runs during re-validation, and biological indicators are typically less expensive than boxes of products. The primary cost concern for re-validation is any product that must be scrapped. Therefore, many companies will accumulate dunnage (i.e., empty packaging or scrap product) over time to fill a sterilizer. This dunnage may be used to ensure that every load is full, or it may only be used for re-validation.
Another alternative to using dunnage for re-validation is to validate a rework process. Any product exposed to a fractional or half-cycle can be re-sterilized in a full cycle. To justify the commercial use of that product, a company needs to validate that the product will not be damaged by exposure to two full cycles. One of the key acceptance criteria for rework is the EO residual levels in the product. However, the manufacturer also needs to determine if any product deterioration by a second exposure to EO would affect performance.
Other EO sterilization considerations
Many companies do a poor job of reviewing the potential impact of changes to a product, packaging, and biological indicators. Ideally, initial validation involves different lots of product, packaging, and biological indicators to assess lot-to-lot variability. However, the packaging and biological indicators often consist of only one lot during validation. Minor changes to the tolerances may reduce the amount of ethylene oxide that is absorbed by the product or change the resistance of the biological indicator to the sterilization process. Therefore, these minor changes should trigger a re-validation.
Changes in suppliers with the same specification can also be difficult to evaluate. If a component is made of a material that absorbs EO, then it may be recommended to re-validate sterilization for any changes to suppliers of those components. Re-validation in these cases may consist of only a fractional cycle, half cycle, or full cycle to evaluate risks associated with the change.
Who should evaluate the need for EO sterilization requalification?
Evaluating the need for re-validation should include inputting three types: 1) microbiological, 2) materials, and 3) performance. To make these assessments, typically, a cross-functional team is needed. Someone responsible for design and development can assess the performance impact of changes. A materials engineer is generally needed to assess the interaction between components and EO. Finally, a microbiologist is needed to confirm that there is no impact related to biological indicators or bioburden.
 Additional Sterilization Training
Additional Sterilization Training
Medical Device Academy has two webinar recordings related to sterilization validation:
If you need assistance with sterilization validation or bioburden failure analysis, please schedule a call with us by visiting our Contact Us page.
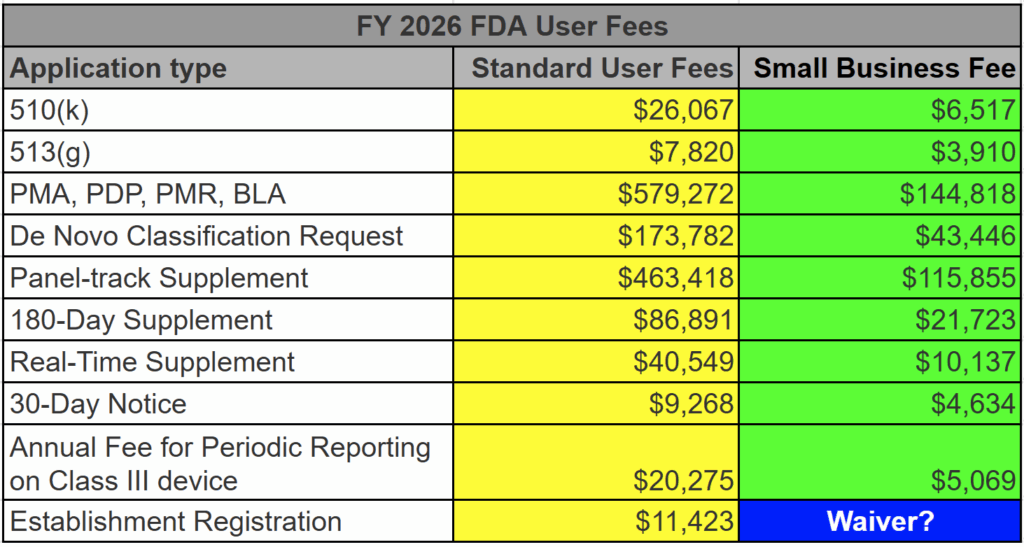
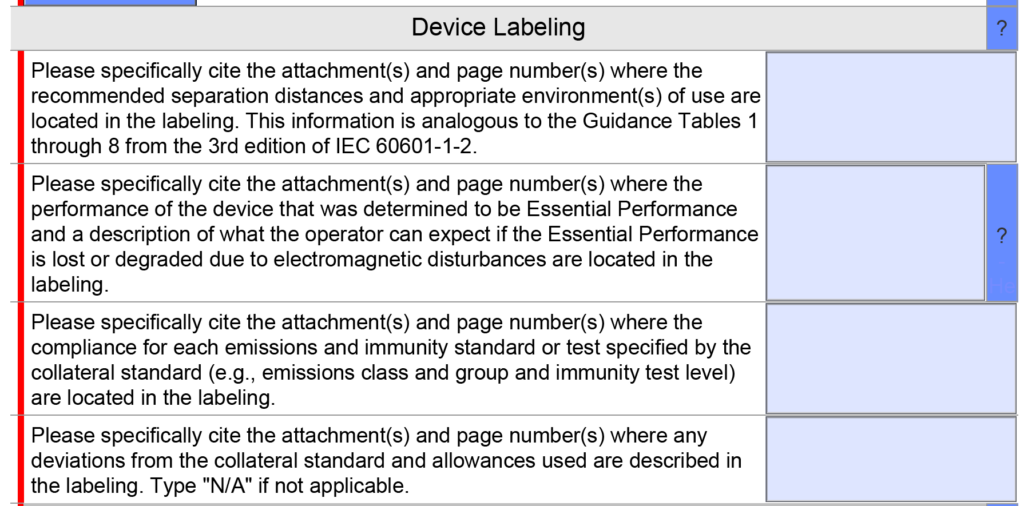

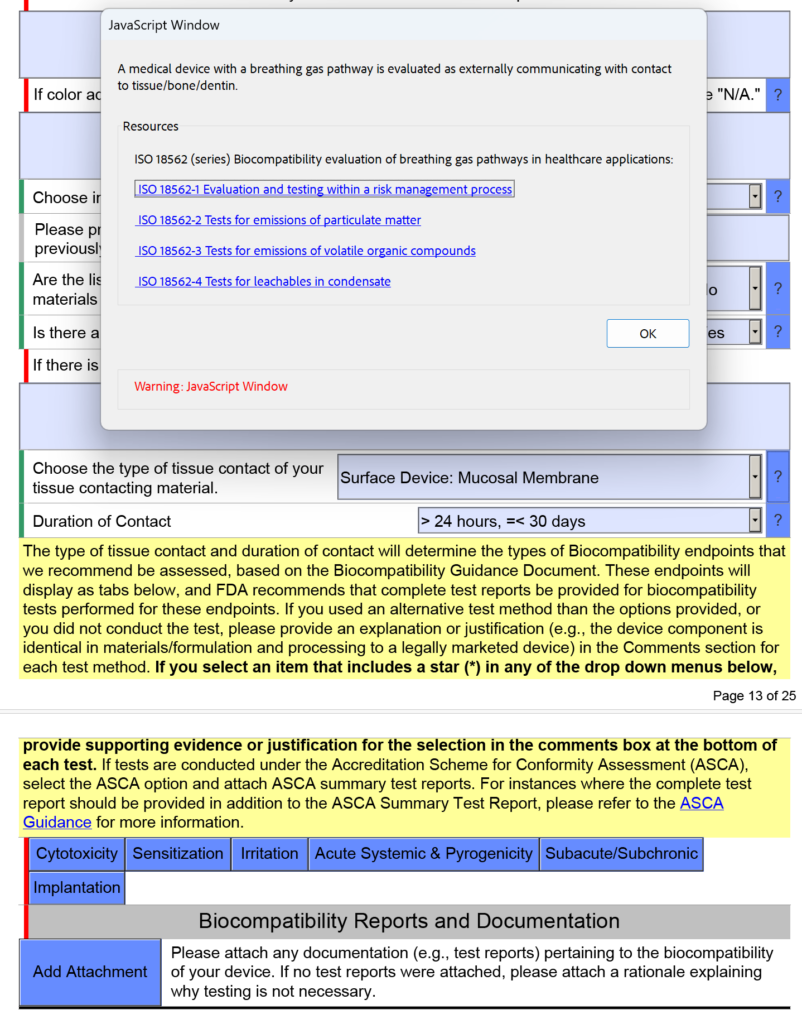
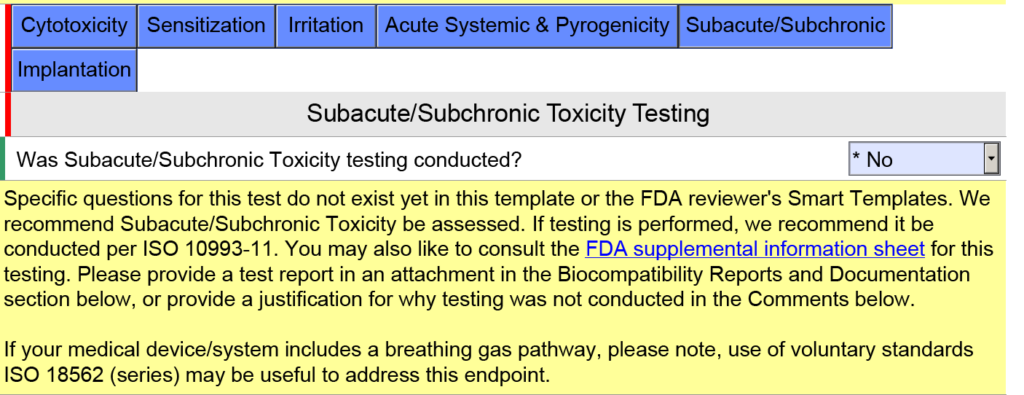
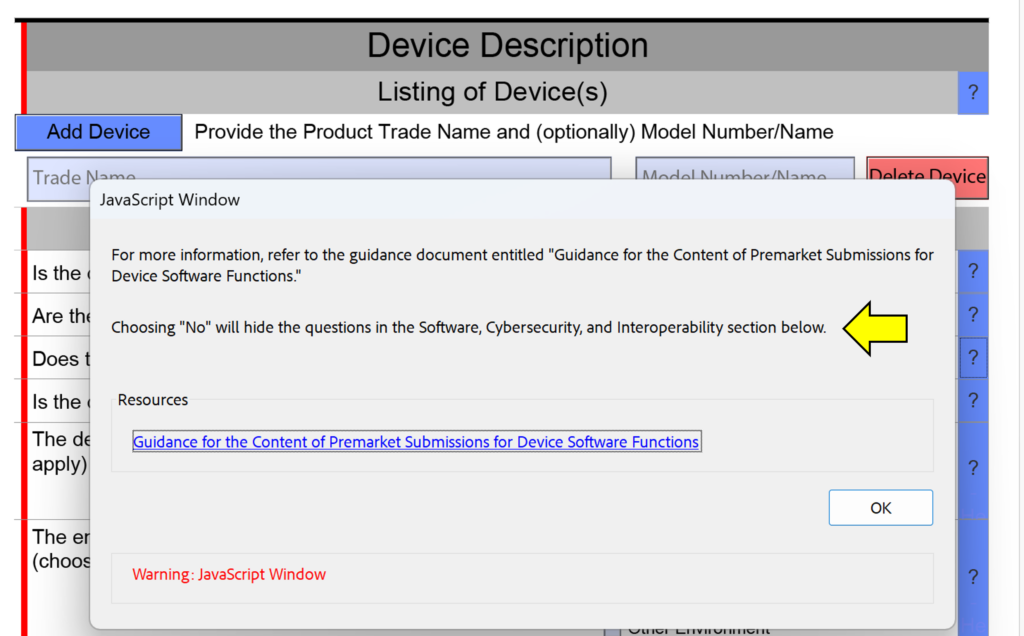
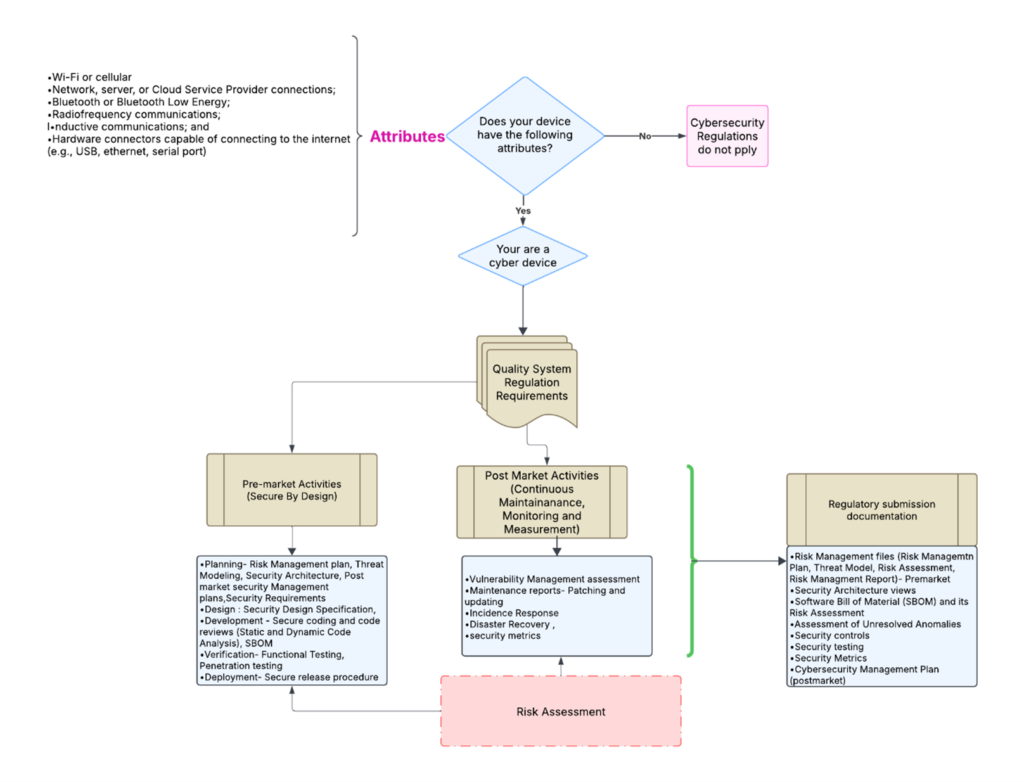
 template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device:
template when this happens. The release of the updated eSTAR version took a little over two months, and the change resulted in a three-page section dedicated to cybersecurity documentation. The previous versions of the template included a requirement for documentation of cybersecurity risk management and a cybersecurity management plan/plan for continuing support. The following documents must be attached in this section if cybersecurity applies to your device: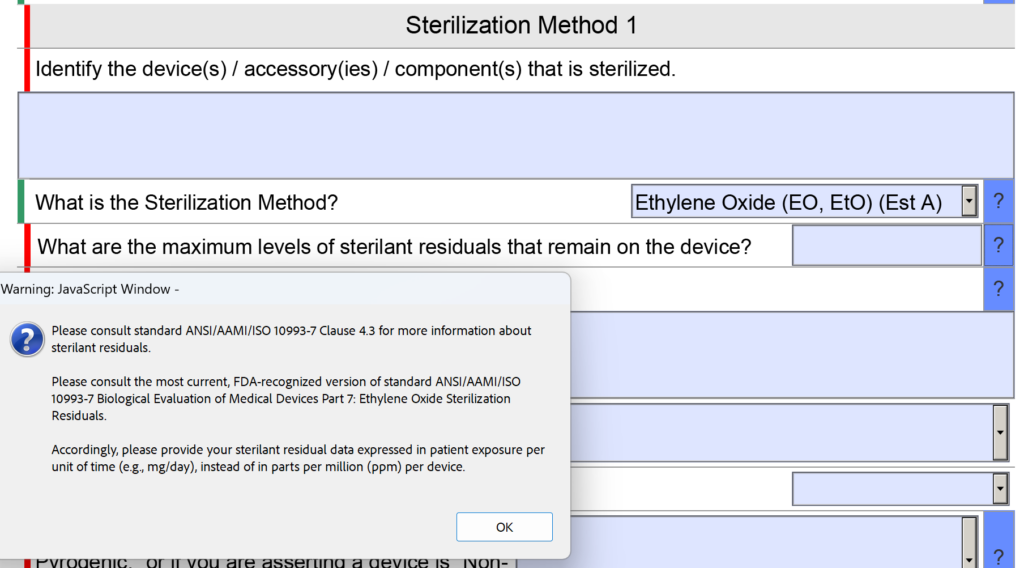




 As the Marketing Lead at BW&CO, Alexina holds a Bachelor of Business Administration in Marketing from the University of Houston. With a deep understanding of the grant funding landscape, she drives strategic marketing initiatives that elevate BW&CO’s brand and reach. Fluent in both English and French, Alexina excels at creating and executing campaigns that connect with diverse audiences. Her expertise includes developing content strategies, managing digital marketing efforts, and conducting market research to support business growth.
As the Marketing Lead at BW&CO, Alexina holds a Bachelor of Business Administration in Marketing from the University of Houston. With a deep understanding of the grant funding landscape, she drives strategic marketing initiatives that elevate BW&CO’s brand and reach. Fluent in both English and French, Alexina excels at creating and executing campaigns that connect with diverse audiences. Her expertise includes developing content strategies, managing digital marketing efforts, and conducting market research to support business growth.