Incoming Inspection – How to perform a single process audit
The incoming inspection process is my favorite process to audit, and it is the best process for teaching new auditors.
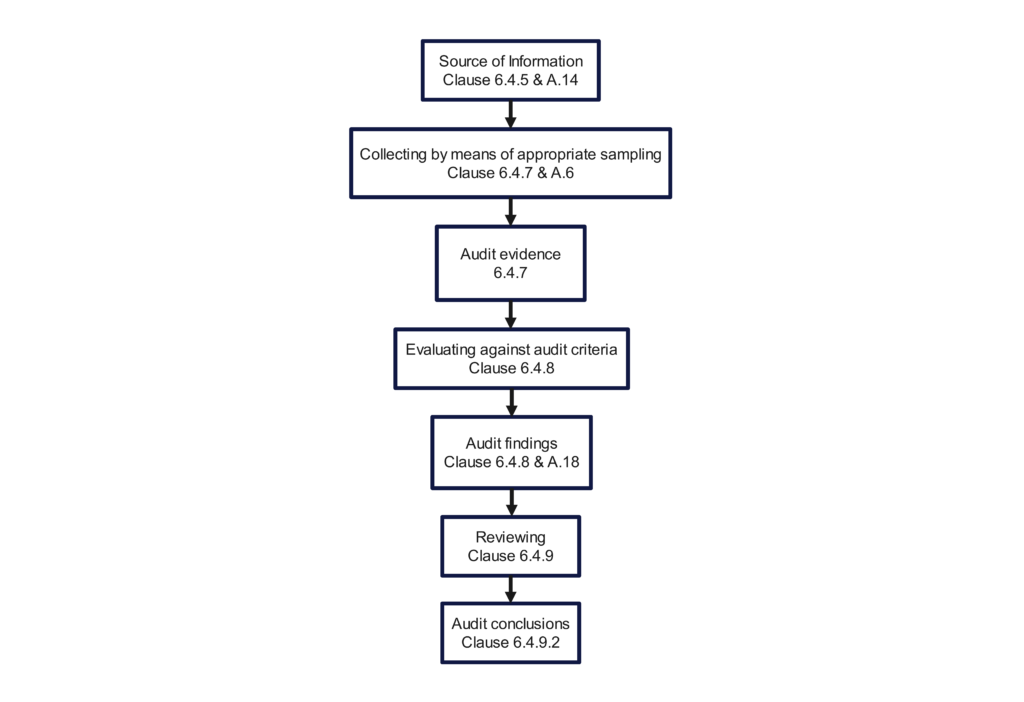
The above video demonstrates how to use a turtle diagram to conduct a process audit of the receiving inspection process. However, this article goes into more detail. You will learn what to look at and what to look for in each part of the audit process.
Preparation for your audit of incoming inspection
If you are conducting an audit of an incoming inspection, you will need a copy of the procedure (i.e., Receiving Inspection Procedure, SYS-033).


Do you need an opening meeting?
Opening meetings are not required for first-party (i.e., internal) and second-party (i.e., supplier) audits. Only third-party auditors are required to have a formal opening meeting. Having an opening meeting is always a good idea, but keep it brief and use a checklist. Try to set the tone for the audit with your opening meeting. This will be your second impression because you already had a conversation with the process owner in preparation for the meeting. However, you want to give everyone present for the opening meeting that you exhibit all the personality characteristics of a good auditor as defined by ISO 19011:2018. Professionalism, organization, and integrity should be obvious to everyone in the room. However, don’t forget to smile and be polite because your auditee might be very nervous. FDA inspectors seem to have an unwritten rule book (i.e., in addition to QSIT) that encourages them to intimidate the companies they inspect.
Step 1 – “Briefly, please describe the incoming inspection process.”
The purpose of this section is not to duplicate the level of detail found in the procedure. It is meant to provide a brief description of the process. Ideally, you want to write a single sentence for the incoming inspection process’s what, where, when, who, and how. A maximum of five sentences is needed to answer those five questions. The process owner should provide the description, and there is no need for them to go into extreme detail because you have at least six more questions to ask (see steps 2-7 below). If you are doing a supplier audit or an audit of a company you don’t work for, you might want to have a few “ice breaker” questions that precede this question. For example, you might ask the person’s name, title, and the number of years they have worked for the company. You might also consider stealing my favorite auditor disclaimer, “If you see me writing furiously, don’t worry. I’m required to write down objective evidence supporting conformity with requirements. If I start asking the same question three different ways, and I’m not writing any notes, that means I am having trouble finding evidence of conformity, and I need your help.”
Step 2 – “What are the inputs that trigger incoming inspection?”
Inputs and outputs of any process refer to both information and physical items. For 100% administrative processes, you may not have any physical items. Incoming inspection, however, has physical goods you receive from suppliers and inspecting. Therefore, the process inputs you are looking for are physical goods and quality system records associated with those goods. For example, if a bunch of titanium round bars were ordered by a buyer in your purchasing department, the physical goods are the titanium bars. The purchase order is one of the quality system records. Other input records that are usually requested to be shipped with the titanium include a packing slip, a certification of analysis, and a dimensional inspection report. It is common to see the incoming inspection activity be delayed because the records are not included with the shipment from the supplier. One recommendation for a process improvement is to require the supplier to send records electronically at the time of shipment instead of sending hardcopies with the product. Statistical inspection sampling plans and work instructions are often confused with input records. These documents are needed to start the incoming inspection, but these are documents that belong in step six of the turtle diagram.
Step 3 – “What are the outputs of the incoming inspection process?”
After incoming inspection is completed there is a requirement to identify the status of the physical product (i.e., accepted or rejected). Usually, a green tag will be used to identify the product as accepted. The tag will also identify the part number, lot, and quantity of product accepted. If the product is titanium, each bar will get a tag. The product will then be transferred to a designated storage area. If you are conducting an audit of a supplier, or a full quality system audit, auditing the warehouse for storage and handling processes is a logical next process. The auditor should look for whether product is segregated in designated locations for specific types of product or if the storage locations are “random” but identified electronically in a material resource planning (MRP) system. The quality system records output from the incoming inspection process will be inspection records and either a green release tag or red rejection tag. If the product is rejected, the product shall be transferred to a quarantine area for nonconforming product and a nonconforming material record (i.e., NCMR) is initiated. Therefore, the process for controlling nonconforming material is another process that could be a logical next process to audit.
Step 4 – “What resources are needed for this process?”
This part of the process approach to auditing is one of the most neglected parts of the quality system. Resources include the facility infrastructure, manufacturing equipment, measurement devices used for inspection, and quality system software used to maintain records of incoming inspection. In this part of the process audit the auditor must be observant. Maintenance records might be located on the side of equipment and they can be reviewed as the auditor walks through the area. This would be an opportunity to interview personnel to make sure they can explain the maintenance process and the equipment maintenance is being performed as planned. The auditor should also determine if equipment validation is required. If the equipment is automated (e.g., automated optical inspection), then an installation qualification (i.e., IQ) should be requested as a quality system record to review at the end of the process or as part of the process for process validation. If the inspection area includes a metrology lab, then the environment may be temperature and humidity controlled. In these types of environments, records of environmental monitoring and trending of environmental conditions should be verified. Lighting, magnification, and particulate filtration could be other environmental requirements for the inspection area. Pest control should be verified in the receiving area, inspection area, and storage areas. The receiving area and warehouse storage are common areas to find pests. Calibration identification should be recorded as a potential follow-up trail for any measurement devices used in the inspection area, and if software is used you will want to verify that quality system software tool validation has been performed.
Step 5 – “Who performs this process?”
A combination of three different roles and responsibilities are typical for this process: 1) department manager, 2) receiving personnel, and 3) inspection personnel. Sometimes one or more of these roles will be combined into one job. The activities sometimes are only performed for a few hours each day, and the personnel that perform the incoming inspection process are assigned to other roles, such as warehouse storage, handling, and shipping. Auditors should always try to interview one or more of the people doing the receiving and inspection activities instead of limiting the interviews to the process owner. Often I will ask the personnel to demonstrate the receiving process and the inspection process. In order to make sure this is possible, you will need to communicate that you want to observe these activities prior to the audit or during the opening meeting. If you don’t, the receiving and inspection activities may already be completed before you start to interview the personnel. Any personnel that are unable to explain the tasks they perform may be targets for verification of training records, effectiveness of training, and competency.
Step 6 – “How is this process performed?”
If an auditor interviews personnel, most people will describe the process in a very haphazard way and steps will be missed. This is why asking people to demonstrate the process is better. The best method is for the person to access the current, approved work instruction or procedure for the process. Then the person should follow the work instruction step-by-step. This allows the person to use the work instruction or procedure as a “crutch” and reduces their nervousness. This also eliminates the skipping steps if the procedures and work instructions are sufficiently detailed. Any blank forms used and statistical inspection standards are also considered quality system documents that define how the process is performed. Sometimes the process owner will provide these documents during their interview, and other times this documents are provided as audit preparation documents. If the documents are not provided in advance the auditor should make sure that they review the documents during observation of activities being performed. This is where an auditor may identify the use of obsolete quality documents, missing details in the documents, and details that are inconsistently followed by personnel.
Step 7 – “What metrics are important for this process?”
Whenever I ask, “What metrics are important in this process?” I typically get a blank stare. Hundreds of business management leaders subscribe to the concept of “what gets managed gets done.” You are also required to establish metrics for your quality system processes in accordance with Clause 8.2.5. Therefore, you need to establish at least one metric, if not more than one. Auditing can help identify opportunities for improvement (OFI), but metrics are the best source of OFIs for a quality system.
Do you need a closing meeting?
You should always conduct a closing meeting for your audits. However, it is also a best practice to summarize your findings for the process owner before you move on to the next process. If some records remain to be reviewed, ensure the process owner knows that the audit results are pending an outcome of reviewing the remaining records. Consider adopting the “sandwich” approach to presenting your findings: 1) something positive, 2) any nonconformities, and 3) something positive. The approach sandwiches the “bad news” between two pieces of “good news.” If you are working as part of a team, the lead auditor should always be aware of the results of your audit. The manager responsible for the process (i.e., the process owner) should also be aware of the results. Do everything you can to prevent unpleasant surprises at the end of the audit.
When you describe any nonconformities, make sure that you include all of the following information:
- the grading of the finding (i.e., MDSAP scoring or Major/Minor)
- a single sentence stating the finding
- the requirement, including a reference to the applicable regulation or standard
- objective evidence from your notes
Whenever possible, email a draft of the wording for your nonconformities to the process owner so they can be prepared with clarification questions during the closing meeting. Make sure you agree with your lead auditor before sending the wording of the finding, and copy them on the email communication. If the process owner has initiated immediate corrective action(s), make sure you note this in your report.
Finalizing your audit report
If you are conducting a supplier audit, you need to give the supplier formal feedback from the audit. You will need an audit report for your quality system records, but you are not required to give the supplier the full report. You might provide a summary of the audit for the supplier instead. If you do this, you should include a copy of that communication in your quality system record (e.g., an appendix to your audit report). If you are going to provide a summary of findings, the content should include at least the following:
- positive findings (i.e., strengths)
- negative findings (i.e., weaknesses)
- nonconformities (if any)
- required actions (e.g., supplier corrective action plan)
- due date(s) for objective evidence of containment, corrections, and corrective actions
- recommendations for follow-up (e.g., next audit)
If you prepare an internal audit report, all of the above content should be included. However, the report should have additional details:
- audit purpose
- audit scope
- audit date(s)
- audit criteria
- name of participants
- date of report
- closure of previous audit non-conformities
- reference to the audit agenda
- deviations, if any, from the agenda
- summary of the audit, including any obstructions
- objective evidence sampled (i.e., what you looked at and what you looked for)
- opportunities for improvement (if any)
Incoming Inspection – How to perform a single process audit Read More »