Additional Information Request is different from an RTA
A poor RTA response will cause a two-week delay, but an additional information request only gets one chance to avoid the dreaded NSE letter.
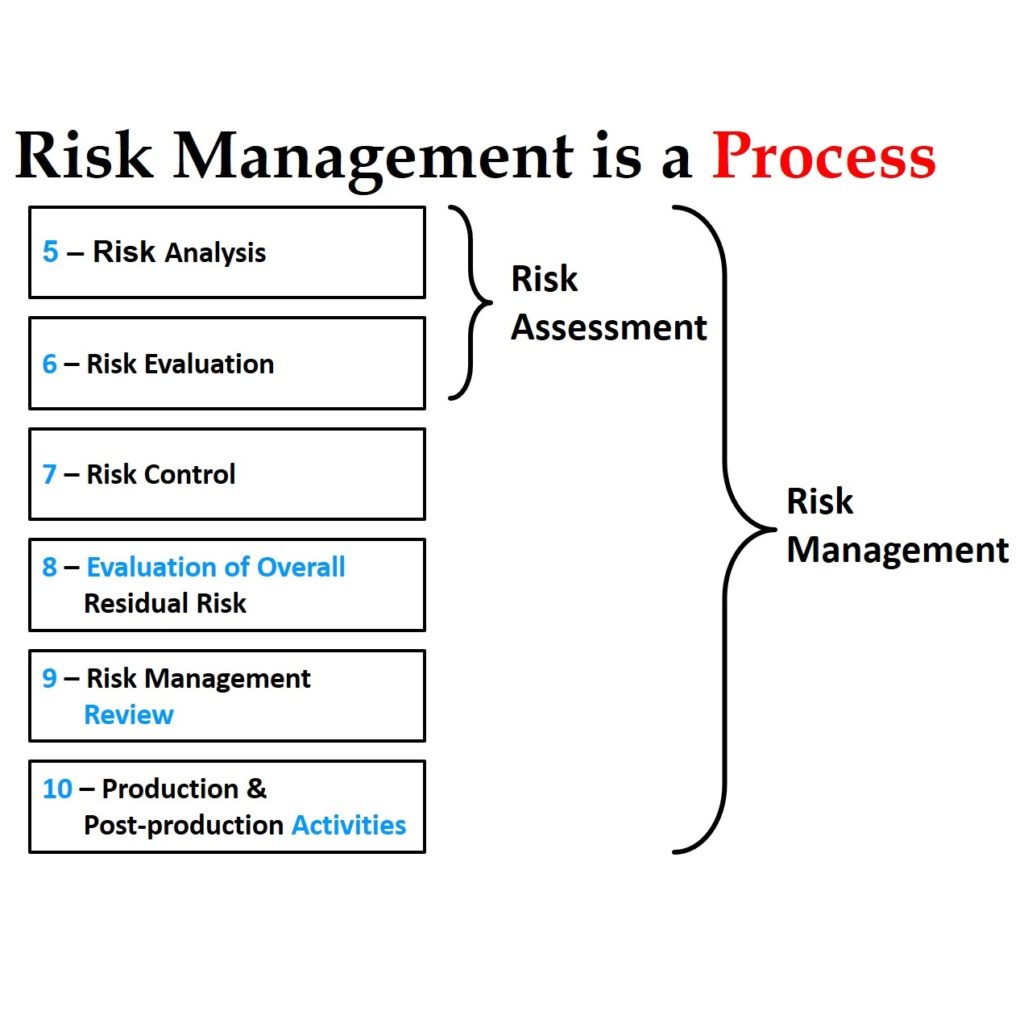
An Additional Information Request (i.e. AI Request) is typically received just before the 60th day in a 90-day 510k review, while a Refusal to Accept (RTA) Hold is typically received on the 15th day. If your response to your first RTA Hold (i.e. RTA1) is inadequate, the reviewer will issue another RTA Hold letter (i.e. RTA2) and your 510(k) review clock will be reset to 0 days. You will have another 180-days to respond to RTA2, and issues identified in an RTA Hold are usually easy to address. Most RTA Hold issues also have one or more guidance documents that are available to help you to obtain an RTA Accept letter. You can always request a submission-in-review (SIR) meeting to clarify what information the reviewer needs to address the RTA deficiencies too. If you want to learn more about responding to an RTA Hold, please read last week’s blog. The rest of this article is specific to responding to requests for additional information.

What happens after 60 days during a 510k review?
On the 60th day of the 510k review clock, or a few days prior to the 60th day, the lead reviewer must determine if they need to issue an Additional Information (AI) Request. The alternative to an AI Request is for the lead reviewer to issue a letter indicating that you have entered the Interactive Review Phase. This only happens if the reviewer believes they can make a decision regarding substantial equivalence in the next 30 days. If the decision is to issue an Interactive Review Letter, then the lead reviewer believes that only minor issues remain and there is only the need for interactive email responses between the lead reviewer and the submitter. An interactive review is the ideal outcome of the substantive review process but it rarely happens.
If you receive an Additional Information Request, what are your options?
The AI letter will indicate that you have 10 days to request a clarification meeting with the reviewer. The wording of this section of the AI letter is provided below:
“FDA is offering a teleconference within 10 calendar days from the date on this letter to address any clarification questions you may have to pertain to the deficiencies. If you are interested in a teleconference, please provide (1) proposed dates and (2) a list of your clarification questions via email at least 48 hours before the teleconference to the lead reviewer assigned to your submission. We would like to emphasize that the purpose of the meeting is to address specific clarification questions. The teleconference is not intended for the review of new information, test methods, or data; these types of questions could be better addressed via a Submission Issue Q-Submission (Q-Sub). For additional information regarding Q-Subs, please refer to the Guidance for Industry and FDA Staff on Medical Devices: Requests for Feedback and Meetings for Medical Device Submissions at https://www.fda.gov/media/114034/download.”
If you wait too long to request the teleconference, then FDA will require you to submit a formal pre-sub request or “Submission in Review” (SIR) meeting request. If you request a SIR meeting within 60 days of receiving an AI Request, the FDA will schedule a SIR meeting with you within three weeks of receiving the request–assuming resources are available. If you wait longer than 60 days to request the SIR meeting, then the FDA will default to their normal target of 60-75 days for scheduling a pre-sub meeting. For example, if you submit your SIR meeting request on day 75, and the FDA takes 75 days to schedule the meeting, you will be granted your SIR meeting at 150 days and you will only have 30 days remaining to respond to the AI Request before your submission is automatically withdrawn.
Therefore, it is important to request a clarification meeting immediately after you receive the AI Request. While you are waiting for your clarification meeting, you should immediately begin preparing any draft testing protocols that you want the FDA to provide feedback on during a SIR meeting. Then after you have the clarification meeting, you should submit your SIR meeting request and include any draft testing protocols you have prepared. This may include a statistical sampling rationale, a proposed statistical analysis method, a summative usability testing protocol, or a draft protocol for some additional benchtop performance testing. The FDA can review examples of preliminary data, a protocol, or a proposed method of analysis. The FDA cannot, however, provide a determination of substantial equivalence.
The Most Common Mistakes in Responding to an Additional Information Request
Most companies make the mistake of asking the lead review if they provide specific additional information, “Will this be sufficient to obtain 510(k) clearance?” Unfortunately, the FDA is not able to provide that answer until the company has submitted the additional information and the FDA review team has had time to review it thoroughly. This is done only when the submitter delivers an FDA eCopy to the Document Control Center at CDRH, and the review team is able to review the information. This new information is assigned a supplement number (e.g. S001), and it will typically require three weeks to review the information. Then the lead reviewer may request minor modifications to the labeling, instructions for use, or the 510k summary. This request is an interactive request, and the submitter must respond within a very short period (e.g. 48 hours), and the wording of the request may be “Please provide the above information by no later than COB tomorrow.”
FYI: “COB” means “close of business.” Wow. The FDA loves acronyms.
Best Practices in Responding to an Additional Information Request
If you receive an AI request on a Friday afternoon, 58 days after your initial submission, you should immediately request a clarification teleconference with the FDA reviewer for the following week. The only exception is if you only have minor deficiencies that you feel are completely understood. During the days leading up to the clarification teleconference, your team should send a list of clarification questions to the lead reviewer and begin drafting a response memo with a planned response to each deficiency. After the clarification meeting, you will have approximately 6-7 weeks to submit a SIR meeting request. However, you should not wait that long. Your team should make every effort to submit your SIR meeting request within 2-3 weeks. If the FDA takes 3 weeks to schedule your meeting, then you will have used approximately 6 weeks of your 26 weeks to respond to the AI Request.
In your SIR meeting request, you should always try to provide examples or sample calculations to make sure the FDA review team understands what you are proposing to submit in your supplement. For example, the FDA reviewers do not have enough time to review your entire use-related risk analysis (URRA) in a SIR meeting request. However, you can provide an example of how you plan to document a couple of use-related risks. Then you can show how these risks would translate into critical tasks. Finally, you could provide a draft summative usability testing protocol for FDA feedback. The FDA review team doesn’t have enough time available to review much more. You will only have one hour for your SIR meeting.
How to Prepare Your Response
In section “V” of the FDA guidance on deficiency responses, the FDA recommends that you restate the issue identified by the reviewer in your response. Next, your response should include one of the following:
- the information or data requested, or
- an explanation of why the issue is not relevant, or
- alternate information with an explanation of why the information you are providing addresses the issue.
Before you respond to an AI Request, you should look up any FDA guidance documents referenced in the AI Hold letter to make sure that you address each requirement in the applicable FDA guidance document(s).
The most important technique to learn when you are responding to regulators is to organize your response in a tabular format that is numbered in exactly the same order that the request was made. Typically there will also be sub-parts to certain issues. In that case, you should duplicate the numbers and/or letters of each sub-part and segregate each sub-part in a different row of the table. Personally, I like to alternate the color of the font I use in the table to make it even more obvious which information is a restatement of the reviewer’s comment and which information is the company’s response to the AI Request.
Why you don’t get a second chance to respond to an AI Request
Once you respond to an AI Request, and the DCC receives your FDA eCopy, the FDA review clock will then resume the countdown to 90 days. In our example above, you received the AIR Request on the 58th day. The FDA must review everything you submitted and make a final substantial equivalence decision before the 83rd day because they still need to submit their recommendations to senior management in their branch. If any changes to the labeling, instructions for use, or the 510k are required, you should receive those requests several days before (i.e. 76-83 days). You can respond to interactive requests via email, and then the final SE decision will be made. If you do not respond to all of the deficiencies in the AI Request, the FDA reviewer will not have enough time to request that you address the remaining gaps and finish their review. Therefore, an incomplete AI Response will certainly result in a non-substantial equivalence (NSE) letter.
If you need to respond to an additional information request from the FDA reviewer, we can review your planned response to identify potential gaps. If you need help please use our calendly app to schedule a call with a member of our team.
About the Author

Robert Packard is a regulatory consultant with 25+ years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Robert was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certification. From 2009-2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Robert’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510(k) submissions. The most favorite part of his job is training others. He can be reached via phone 802.258.1881 or email. You can also follow him on Google+, LinkedIn or Twitter.
Additional Information Request is different from an RTA Read More »