This article explains the FDA regulations related to private labeled devices that are already 510k cleared and distributors want to import.

This article was initially inspired by a question asked on the Medical Devices Group website hosted by Joe Hage. Companies often ask about how to private labeled devices in the USA, because they are unable to find anywhere in the FDA regulations where private labeling of the device is described. The reason for this is because the FDA regulations for devices allow for the labeling to identify the distributor only—without any mention of the OEM manufacturer on the label. In contrast, most other countries have “own-brand labeling” regulations or regulations for private labeling devices. It is also important to remember that the FDA only approves devices through the pre-market approval (PMA) pathway. All other devices fall into one of three categories: 1) 510k exempt, 2) 510k cleared, or 3) De Novo classification request approved. Devices that fall into the third category will subsequently fall into category 1 or 2 after the FDA approves the classification request.

Questions about the private labeled devices process for FDA
Our distribution company is interested in getting a private labeled devices agreement with an OEM to sell a Class II medical device in the USA. The OEM has 510(k) clearance, and the only product change will be the company’s name and address on the label. There will be no change to the indications for use. Please answer the following questions:
- Is it legal to eliminate all mention of the OEM from the device labeling?
- Who is responsible for complaint handling and medical device reporting? OEM or private-labeled distributor?
- What is the process to get this private label for the Class II device?
- How can our distribution company avoid paying the FDA user fee?
Answer to the first question about private labeled devices
The FDA is unique in that they allow either the distributor or the manufacturer to be identified on the label, but both are not required. Therefore, if Joe Hage were the distributor, and you were the manufacturer, there are two legal options for the private labeled device: 1) “Distributed by Joe Hage”, or 2) “Manufactured for Joe Hage.”
The manufacturer is not required to be identified on the label. However, the OEM must be registered and listed with the FDA. If the OEM is outside the USA, then the distributor must register and list with the FDA as the initial importer and reference the K number when they complete the FDA listing. There is no approval required by the FDA. You will need a quality agreement defining the roles and responsibilities of each party, but that is all.
Answer to the second question about private labeled devices
The quality agreement must specify which company is responsible for complaint handling (21 CFR 820.198) and medical device reporting (21 CFR 803). In this situation, the OEM is the specification developer, as defined by the FDA. Therefore, the OEM will be responsible for reporting and execution of recalls. Therefore, even if the distributor with a private label agreement is identified as the “complaint file establishment,” the OEM will still need to obtain copies of the complaint information from the distributor, and determine if medical device reporting and/or corrections and removals are required (i.e., recalls).
Answer to the third question about private labeled devices
There is no formal process for “getting a private label.” The entire private label process is negotiated between the distributor and the OEM with no involvement of the FDA. However, in the listing of devices within the FDA FURLS database, all brand names of the device must be identified. Therefore, the OEM will need to add the new brand name used by the distributor to their listing for the 510(k) cleared product. However, the FDA does have the option to keep this information confidential by merely checking a box in the device listing form.
Answer to the fourth question about private labeled devices
If the distribution company is the initial importer of a device into the USA, then the distributor must be registered with the US FDA as the initial importer, and the distributor will need to pay the FDA user fee for the establishment registration. That user fee is $5,236 for FY 2020, and there is no small business discount for this fee. The only way to avoid paying the user fee is to have another company import the device, who is already registered with the FDA, and to distribute the product for that company. I imagine some logistics brokers might be acting as an initial importer for multiple distributors to help them avoid paying the annual FDA user fee for establishments. That company might also be providing US Agent services for multiple OEMs. However, I have not found a company doing this.
Is private labeling of device legal in the USA?
The FDA is unique in that they allow either the distributor or the manufacturer to be identified on the label, but both are not required. Therefore, if Joe Hage were the distributor, and you were the manufacturer, there are two legal options for the private label: 1) “Distributed by Joe Hage”, or 2) “Manufactured for Joe Hage.”
Who must register, list, and pay user fees for medical devices?
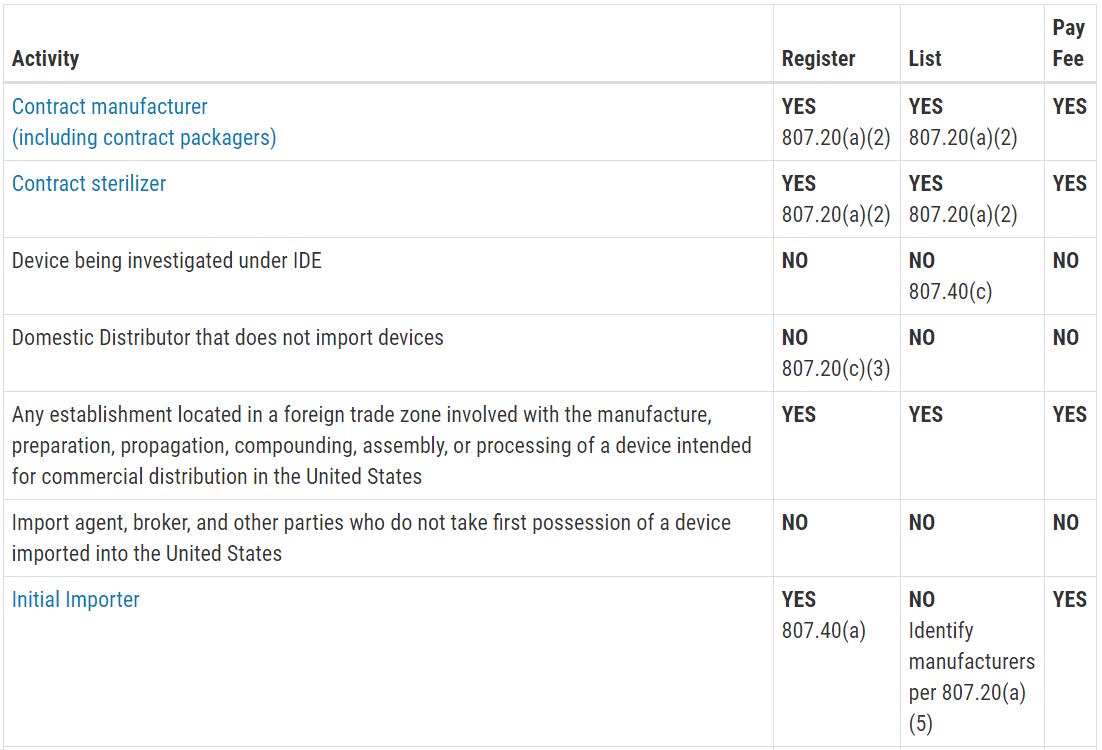
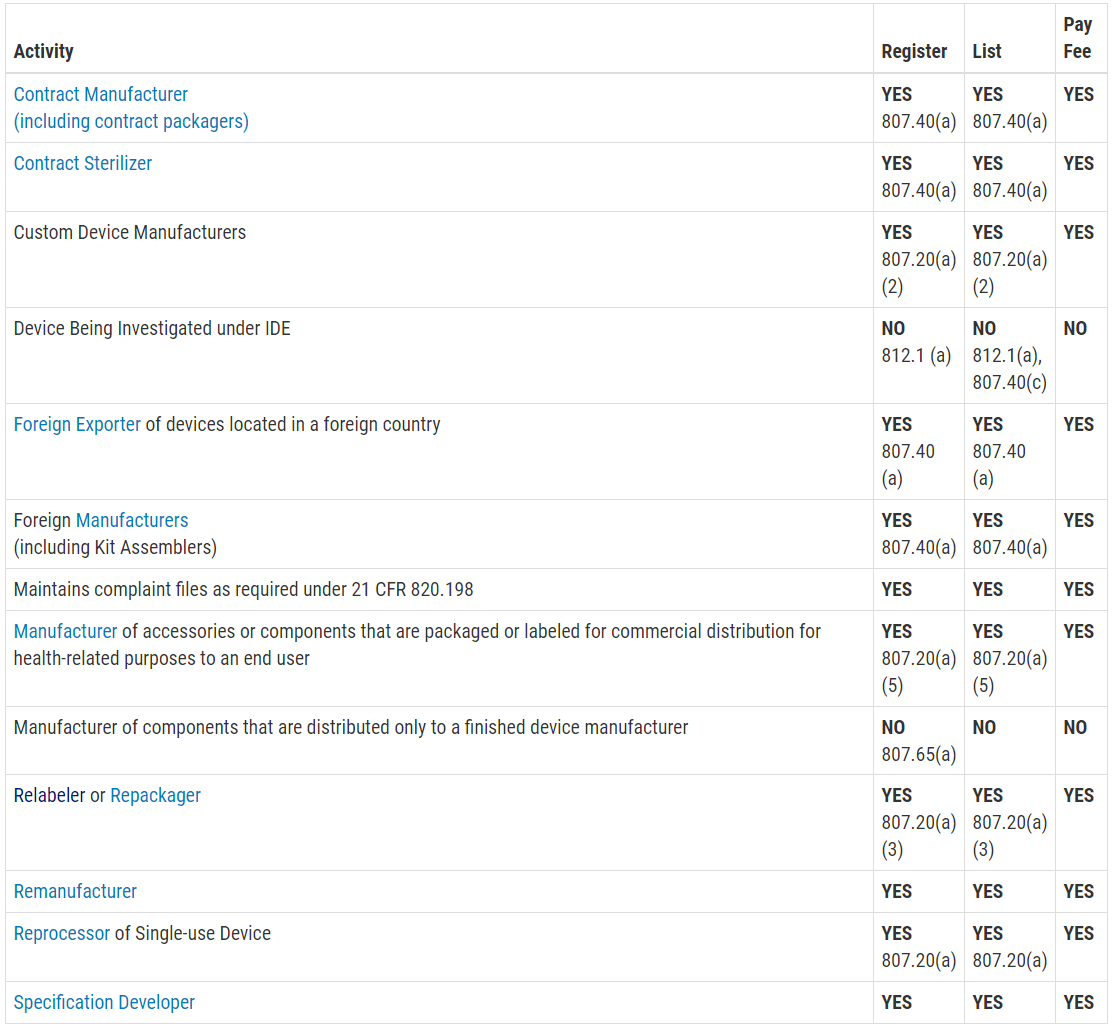
This question is frequently asked, and the table with the information was not visible on my mobile browser. Therefore, I copied the table from the FDA website and posted the information in the image below. The information is copied directly from the FDA website:
Registration and Listing Requirements for Domestic Establishments

Registration and Listing Requirements for Foreign Establishments
For products that are manufactured outside the USA, and imported into the USA, the initial importer is often the company identified on the label. There are two typical private labeling situations, but other possibilities exist:
- If the initial importer owns the 510(k), then the manufacturer outside the USA is identified as the “contract manufacturer,” and the initial importer is identified as the “specifications developer.” Both companies must register their establishments with the FDA, and there needs to be a quality agreement between the two companies defining roles and responsibilities. The contract manufacturer outside the USA is not automatically exempt from reporting requirements and complaint handling. The contract manufacturer outside the USA may decide to label the product as a) “Manufactured by”, b) “Manufactured for”, or c) “Distributed by.” Options “a”, “b” and “c” would list the importer’s name because they own the 510(k), and they are the distributor. This situation often occurs when companies outside the USA want to sell a product in the USA, but they do not want to take on the responsibility of obtaining 510(k) clearance. These firms often believe this will exempt them from FDA inspections, but the FDA is increasingly conducting FDA inspections of contract manufacturers due to this private label situation.
- If the manufacturer owns the 510(k), then the manufacturer outside the USA is identified as the “specifications developer” and the “manufacturer,” while the initial importer will be identified as the “initial importer.” The importer may also be specified as the complaint file establishment and/or repackager/relabeler in the FDA registration database. The manufacturer outside the USA will not be able to import the device into the USA without identifying an initial importer in the USA in the FDA FURLS database. The manufacturer outside the USA may decide to label the product as a) “Manufactured by”, b) “Manufactured for”, or c) “Distributed by.” Options “b” and “c” would list the importer’s name, while option “a” would list the manufacturer’s name. This situation often occurs when US companies want to be the distributor for a product made outside the USA, and the company wants a private labeled product. This also happens when the OEM wants the option to have multiple US distributors.
In both of the above private-label situations, the non-US firm must have a US Agent identified because the company is located outside the USA. The US Agent may be the initial importer, but this is not required. It could also be a consulting service that acts as your US Agent. The US Agent will be responsible for receiving communications from the FDA and confirming their role as US agents each year when the registration is renewed. Medical Device Academy offers this service to non-US clients we help obtain 510(k) clearance.
Follow-up questions
A Korean company, with a US distribution subsidiary, would like to private label a medical device with an existing 510(k) owned by another company in their name. Does the Korean company need a contract in place before private labeling? Does the US subsidiary and/or the Korean parent company need to be registered in the USA prior distribution of the private-labeled version of the device in the USA?
Rob’s response: Initially, it was unclear from the wording of the question as to whom is the 510(k) owner, which company will be on the label, who is doing the labeling, and who is doing the importing to the USA. The person asking Joe Hage this question tried clarifying their question via email, but we quickly switched to scheduling a phone call using my calendly link. I have reworded the question above, but here are some of the important details I learned during our phone call:
- The person asking was already acting as the relabeler, repackager, and they were distributing the product in the USA. This person’s company is also registered with the FDA.
- The device is 510(k) cleared by another US company, and there is no need to worry about the complications of an initial importer being identified for a product manufactured in the USA.
In this situation, the relabeler/repackager can relabel the product for the Korean company’s US subsidiary as long as there is a quality agreement in place for all three parties (i.e., relabeler, distributor, and manufacturer). There is no need for the Korean parent company to register with the FDA. There is no need for a new 510(k) submission, and the US subsidiary does not need to register with the FDA—as long as the quality agreement specifies that the US subsidiary will maintain records of distribution, facilitate recalls if required, and notify the manufacturer of any potential complaints and/or adverse events immediately. The manufacturer with 510(k) clearance will be responsible for complaint handling, medical device reporting, and execution of recalls according to the agreement. The relabeler will be responsible for maintaining records of each lot of product that is relabeled for the US subsidiary, and the relabeler must maintain distribution records that link the original manufacturer’s lot to the lot marked on the relabeled product.
If you have questions about the private labeling of your device, please contact us.
The proper Medical Device labeling service is required to control unnecessary human exposure to potentially hazardous ionizing and nonionizing radiation, and to ensure the safe, efficacious use of such radiation.
Hello !
This article is very interesting and full of precious information !
I would like to know if there is any quality agreement template for this typical situation (private labeling with a relabeler).
Thanking you in advance,
Thank you for the excellent suggestion. We have a webinar scheduled for June 25, 2020. I will add the creation of a quality agreement template for relabeling to the agenda. If you want to register, please visit this webpage: https://medicaldeviceacademy.com/suppliers-qualified-prove/
We already have a supplier quality agreement template sold with our supplier quality management procedure (SYS-011): https://medicaldeviceacademy.com/standard-operating-procedures-medical-device-academy/supplier-quality-management-procedure/
Hi Bob,
Nice article.
Just curious to know whether the OEM must have the 510(k) approved for the product before private labeling. or there is an option where the private labeler will take the OEM product brand it in his name , obtain 510(K), register with FDA and place the product in the US Market.
Appreciate your help in advance.
Thank
Thank you for the positive feedback on the article. If the product is a Class 1 exempt or Class 2 exempt device, then a 510(k) is not required by either company. If the product requires a 510(k), then the 510(k) is required before either company can register with the FDA. In the situation you described, the OEM is actually considered a contract manufacturer with the FDA. You would be the specifications developer and initial importer, while the OEM registers as a contract manufacturer. However, neither company can register the product without a K number.
Hi Bob,
This article is very informative and very easy to understand. It answers lots of questions one might face during dealing with listing for the 1st time.
To the third question about private labeled devices, does the OEM need to do add to file due to new brand name used by distributor before adding it to listing of the 510K cleared product?
Thank you in advance.
Thank you for your question.
The OEM needs to update the listing information to indicate the new brad name, and this would also require updating the Device Master Record (DMR) for any labeling if it is done by the OEM for the distributor.
Rob
Hi Rob,
This article is very informative and helpful,
If we are the initial importers of the products(Class I not exempt)based in US. The products are manufactured in Thailand which already hold a 510(k). We want to Relabel/ Re-brand them under our own brand name.
My question is can we hold a separate 510(k) for the products?
Appreciate you help in advance
Thank you
Yes, you can obtain your own 510(k) for the device. However, you are not “relabeling/re-branding” in this scenario. You are becoming the specification developer, and the supplier is the company you are outsourcing design to and manufacturing. You will also have to comply with the current standards and testing requirements. You may be able to provide a biocompatibility certification statement instead of doing your own biocompatibility testing, but you would need documentation from the supplier to support that as well.
Never have I seen such a clear explanation for an FDA regulated process.
Thank you!
Hi Rob,
Thank you for the very informative article.
I do have an additional scenario which I would like to have your insight on:
A non-US company would like to distribute under its brand name a device to the US market. The device is manufactured by another non-US manufacturer (OEM) which own a 510K for the device.
The distributer has a US agent which also acts as an initial importer for other devices.
How to label the device and who is responsible and liable for the products.
Thanks in advance
There should be quality agreements that are established between the OEM, the distributor, and the US agent. The quality agreement should specify roles and responsibilities for each party. Typically a US agent is not an initial importer too, but if the US agent is also providing this service they will need to maintain distribution records, and they will need to be responsible for storage, handling, and shipping of the devices. Usually, the initial importer will require that they are indemnified by under the liability insurance, because they would not have control over design, manufacturing, or labeling of the product.
The OEM must be identified as the specification developer in the registration, because they own the 510(k). They also control the manufacturing of the device. The OEM is probably also applying the labeling for the distributor. Therefore, this organization will typically hold the liability insurance for the product, and they would be the primary establishment for FDA inspections. Depending upon the agreement, they may or may not be responsible for compliant files related to the product with the distributor’s name. But the agreement would state the distributor would communicate complaints they receive to the OEM so the OEM can perform investigations of root cause–and potentially recall product. Since the distributor’s name is on the product, the distributor will need to assign the “DI” portion of the UDI number.
Finally, the distributor is responsible for verifying that the US Agent is maintaining distribution records and maintaining the data in the GUDID. They will probably be identified as the complaint file establishment.
What would be the labeling requirements where the design and specification developer is in the US but has a contract manufacturer doing all of the manufacturing with FG release? Does the CM have to be listed on the label or can only the design/specification developer be listed as the “Manufacturer”? Thank you
The US company can be listed as the specification developer and complaint file establishment, and the CM can be listed as a “manufacturer.” However, the label in the US will then be marked with “Manufactured for” instead of “Manufactured by”.
Hello Rob!
Thank you very much for this great information!
I have a question for you. I am a company based in Canada, and my supplier already has the 501(k) for this medical device.
I want to Private Label this Medical Device and sell it on Amazon in the US.
So Amazon will do all the storage, shipping and handling.
Will it affect me as being the Initial Importer in any way? I will still have to go through the exact same process and laid out in your article?
Thank you very much!
Unfortunately, even though Amazon is the initial importer you will be considered the complaint handling establishment and repackager/relabeler. Even though your supplier has a 510(k) and they will be labeling the product for you, if your name is on the product you will be responsible for 3 things:
1. registration as complaint file establishment
2. registration as repackager/relabeler, because your name is on the label
3. applying for a GUDID account with FDA and assigning the DI portion of the UDI barcode for your product (your supplier will assign the PI portion); this includes uploading the GUDID data elements into the FDA database
Amazon will be the initial importer and they will maintain records of distribution.
As the repackager/relabeler you also need a quality system and you are subject to FDA inspection. Even though your supplier will do most of the work, you still need to control them as a supplier and make sure all of the outsourced activities are being performed. This typically involves customer audits, but this might be a remote audit. You will need to execute a supplier quality agreement with the supplier that articulates roles and responsibilities for both sides.
Rob
Hi great article!
I have a question, maybe mentioned already but could not find it specifically.
Company A is holder of a 510K and is outside of the US, does not do import into the US.
Company B buy’s products from company A outside of the US (does not have an establishment in the US).
Company B is a manufacturer of its own products and has multiple Initial imports for her own products into the US.
Company B wants to sell the product of company A (company A mentioned as manufacturer or private label) to the US using her own initial imports network.
Is this possible without sharing the sales network with company A?
That’s a great question. Yes, Company B can sell the product of company A. There should be a distribution agreement signed between the two companies, and the agreement needs to define who is responsible for the complaint files and medical device reporting for product labeled under Company B’s name. Company B may require Company A to conduct investigations for complaints, and in those cases the customer information will need to be shared. In addition, Company A may need COmpany B to share complaints data in aggregate form for the purposes of post-market surveillance. Finally, one of the two companies will need to label the product with Company B’s name. If B is doing relabeling, then Company B needs to be registered as a repackager/relabeler too.
Hi Rob,
Thanks a lot for the informative article.
I have an additional scenario which I would like to have your opinion on it:
Company A (non-US company) would like to distribute under its brand name a device to the US market. The device is manufactured by company B [non-US manufacturer (OEM)] which do not own the 510K clearance for the device.
In this case, can company A be the holder for 510K and private-label the device?
How to label the device and who is responsible for the products.
Thanks in advance
Hi Andrew,
Thank you for the question. In the scenario you are describing, if Company A submits a 510(k), then Company A becomes the specification developer and Company B becomes a contract manufacturer. In this scenario, Company B would need to share the complete design history file (DHF) with Company A in order to be compliant with the FDA requirements, and Company A would need to maintain a DMR. This can create a problem if there are particulars of the design or manufacturing process that are proprietary. Company A is also no longer considered a private labeler in the eyes of the US FDA. Company A would need to apply for a GUDID account and obtain a UDI for the device, because the name on the label is Company A. Company B would primarily be responsible for the manufacturing activities, while Company A would be responsible for almost everything else–including complaint handling, MDRs, recalls, etc. The labe would say: “Manufactured by Company A”.
Rob
With respect to private labeling. Is there a way in fact to completely hide the identity of the supplier. Or is it that anyone doing their homework can find the supplier. The concern of course are competitors and customers going direct. So how can the source be hidden.
When a company lists a brand name associated with its establishment in the FDA database, there is an option to remain confidential. However, the company is still registered in the FDA database. Only the association with the specific brand name listing is confidential.
Company A is a US based distributor and relabeler/repackager. Company A has a contract manufacturer (Company B) in a foreign country. Company B would like to private label the device for other US distributors. Company B would like to pay a licensing fee to Company A for use of the 510(k). Is this possible, or does Company B need their own 510(k) in order to private label for multiple US distributors?
In the scenario you are describing, Company A is not just a distributor, relabeler/repackager. Company A is also the initial importer, complaint handling establishment, and the specification developer. The reason is that Company A holds the 510(k) and they are located in the USA, while Company B is outside the USA. If another US-based company wanted to be the distributor, then Company A remains the specification developer. However, you would need a 3-way supplier agreement that defines who would be responsible for complaint handling of devices distributed by the new company. Product can be shipped to either Company A or the new company. If product is shipped to company A and complaints are handled by company A, then the new company does not have to register with the FDA either.
Hi Rob,
Thank you for the amazing article!
Does the OEM or Private labeler have responsibility for UDI and GUDID listing?
Thanks
Good question. The company listed on the label is required to apply for the GTIN from the issuing agency and to apply for the GUDID account from the FDA. The FDA will not give another company the account–even if you are outsourcing the labeling creating and control to that company. If you private label you will need to give the prefix numbers to your supplier and you will need to enter information in the GUDID.
Your article was very informative.
Company A is a non-US company that has 510k cleared products and is fully registered with the FDA. Company B would like to sell Company A’s products in the US market and on Amazon under Company B’s own name on the label. Is it possible for Company B to do so without registering with the FDA and paying the associated fees?
I would greatly appreciate your valuable feedback on this. Thank you.
Great question Naveed. The short answer is no.
In your example, Company B is an initial importer, Company B is a repackager/relabeler, Company B needs a UDI account, and Company B needs a quality system. Company B might also need to be the complaint file establishment–depending upon the quality agreement between Company B and A.
Company A will be registered as the specifications developer and probably the manufacturer. Company A will also need an FDA US Agent.
Thanks for this info! Quick question. If a US based clinic has created a patented Class I device and is working with three manufacturers in China to have it made and assembled, do all the manufacturers involved need to be FDA registered or is it enough to just have one part made by a manufacturer with FDA registration?
Thanks!
Many companies think they are not required to register with the FDA as a contract manufacturer for various reasons. If your company is a contract sterilizer or your company is making a finished device, you must be registered–regardless of where you are shipping it initially. In your example you mentioned three manufacturers. If they manufacturers are making components, they are not required to register. If they manufacturers are making accessories or finished devices, they are required to register. If two component suppliers ship to a third supplier that does final assembly and packaging, then only the final assembly company is required to register.
Hi Rob,
Is it acceptable to have manufactured for ‘ ‘ and distributed by ‘ ‘ on the labeling? The name in quotes would be the same for both, this would save a company from having to label EU product and US product separately but I don’t know if it’s an acceptable practice. Thanks!
This is not an acceptable way to label the product in the EU. There are official symbols to identify distributors, importers, manufacturers, and authorized representatives. Those symbols need to be used on CE Marked devices. The FDA allows you to use these symbols as well, because they recognize ISO 15223-1. In addition, private labeling is not permitted in the EU. Therefore, in general the best approach is to rework labeling or to label product specifically for the market you intend to sell it in. The same is true for instructions for use and user manuals. Just because the industry has been able to use the same labeling globally in the past doesn’t mean that we can continue to do that. On-demand labeling will become the norm, and it can prevent many of the labeling nonconformities we see during audits and inspections.
Great article. I have a question for you. If we are private labeling a product from a non-US company and only bringing the products into the US for packaging and labeling but selling to another country and not the US at all (at least for the next few years), do we have to establish ourselves with the FDA and what process do we need to go through with the FDA. TIA
Thank you for the comment on our article. I hope you liked our most recent blog too.
In the situation you described, the company in the US must register as “U.S. Manufacturer of export only devices.” Companies with that role, must register [807.20(a)(2)], list [807.20(a)(2)], and pay a fee.
Pingback: FDA Approval Process: "Triage" for 510(k) - Medical Device Academy Medical Device Academy
Thanks for the interesting article.
I have one question. Company A is the manufacturer of a device. Company A now removes their own brand and labels the device with the brand of Company B and adds the phrase “manufactured for Company B”. Company A adds the new brand name in their FDA listing as manufacturer. Company B does not physically change the labeling and packaging of the device. Is Company B nevertheless considered as relabeler/repackager although they do not physically relabel the device?
Hello, I hope you can help us navigate through this mess.
Neither of the two companies has a license.
Company A is a company based in the United States,
Company B is a private label manufacturer located in China.
Company A is having a Class III medical device manufactured by Company B. However, Company B, as the manufacturer, does not want to obtain a license. In this case, if Company A were to order licenses from both parties, what should it choose, and is it possible?
Another question:
In the same scenario, if these products are manufactured under private label in China and then brought to the United States, and subsequently exported to Canada, what licenses should be obtained?
A US company that is importing a product manufactured outside the USA can serve as the US Agent, but there is no “license.” Instead, the company in China must be registered with the following roles:
1. specification developer
2. manufacturer
The company in the USA must be registered with the following roles:
1. repackager/relabeler
2. initial importer
3. complaint file establishment
Both companies must be compliant with US regulations, including quality system requirements. There also needs to be a supplier quality agreement in place that indicates who is responsible for each requirement and which requirements are shared. The manufacturer outside the USA can choose whomever they wish as the US Agent, but the US Agent must reside in the USA. If the company outside the USA is not willing to register with then then the product will not be permitted into the USA by the FDA. The above rules apply to all device classifications.
Most private labeling of devices for the US market are for Class 1 and Class 2 devices. The reason for this is that pre-approval inspections by the FDA are required for Class 3 devices.
In your follow-up question, you need to determine if the product will be sold in the USA or “for export only.” If the device is for export only, then the company inside the USA needs to be registered with the FDA as “U.S. Manufacturer of export only devices.” I believe that the manufacturer outside the USA will still be required to register as a contract manufacturer, because otherwise the devices will not be admitted through customs, but this scenario is not described in the regulations.
If you would like additional help with registration and/or private labeling requirements, please schedule a call using the calendly links on our Contact Us page.
If the company is considered the specification developer, foreign exporter, complaint handler, and relabeler, how does the FDA annual registration payment work? In terms of FY 2024, would the registration fee be $7653 or $30612 ($7653 x 4 because of the 4 different establishment groups).
User fee is per establishment–not per role. Therefore, the fee is $7,653 for FY 2024 regardless of the number of roles.
Hi Rob,
I recently discovered your company and just wanted to say thank you for providing such great resources!
I did have a quick question. A foreign vendor holds the 510(k) and manufactures the product for my company but with my company’s name on it (“manufactured for” my company on the label). If my company in the US is designing the label and transferring it to them, am I considered the relabeler or a spec developer? I’m designing the label based on what they were cleared for in the 510(k) but I’m just adding logos, SKU and branding for my company.
Alternatively can I say that because I’m designing the label (making the draft in Adobe illustrator) but the supplier overseas approves it and incorporates it into their own DHF/DMR that I am not actually the relabeler or spec developer? I have an FDA registration already but I’m just registered as a complaint file establishment so I’m trying to see if I need to register a different function.
Lastly, in this relationship where I have quality agreement pointing to them as the specification developer, do I need to maintain a DMR with the vendor’s product specifications for can I “point to the location of” all the files per the regulation to just be housed with the supplier?
This particular situation comes up all of the time. Most companies are trying to avoid having a quality system and registering as a repackager/relabeler. The specific responsibilities you have depend on your agreement. If your supplier is reviewing and approving the final label you create, and they add the brand to their device listing, then you are the initial importer/distributor and you are not the relabeler.
The specification developer is the 510(k) holder.
If you would like more specific help for your situation, please schedule a call with me.
Hey Rob,
Thanks for your knowledge sharing. I have a question regarding this article.
We have a manufacturer whose Class 1 devices are listed in the FDA FURLs portal as a generic Manual Surgical Instruments like ‘Retractors’.
We are listed as an importer. Can we use a specific brand name for this item like “ABCRetract Retractor” or our supplier who is the manufacture first need to list this terminology/brandname under his device listing?
FDA customs authorities review the shipping documents to make sure that it matches the devices that are listed with the FDA. Therefore, it is important that your supplier update the listing to include the brand name that you product is labeled as. If this is not done, the product may be impounded by customs.
Hey Rob,
Thanks for your knowledge sharing. I have a question regarding this article.
We have a manufacturer whose Class 1 devices are listed in the FDA FURLs portal as a generic Manual Surgical Instruments like ‘Retractors’.
We are listed as an importer. Can we use a specific brand name for this item like “ABCRetract Retractor” or our supplier who is the manufacture first need to list this terminology/brandname under his device listing? Moreover, will we be able to use our UDIs in this case or from the manufacturer?
I am answering this question separately. The FDA requires that the UDI and GUDID account be in the name of the company designated “UDI labeler.” If your supplier uses their UDI codes, the listing must be recorded in their GUDID account, then your company is the initial importer. However, if you are using your company’s UDI codes, then you must obtain your own GUDID account. In addition, your company will need to register as a repackager/relabeler and you will need a quality system to control labeling review and approval and to maintain the identification and traceability with your UDI barcodes. There are a lot of different variations on this situation and it may change the answers you are given. Therefore, if there is any doubt you should submit a question to the UDI help desk.
If we are a US Mfg for a medical device designs, manufactures, labels and distributes the cleared 510k and also relabels the same device with another company’s name, logo and branding for US distribution as well, are we still the legal manufacturer for the later scenario?
Yes, you are the legal manufacturer. The other company should be identified as “Manafactured for” or “Distributed by.”
As a relabeler, am I responsible for full design control (V&V), generating a risk management file, and/or DHF/MDF for the new label?
As a relabeler you are responsible for maintaining design controls and managing design changes as they pertain to the labeling. You will need an agreement with your supplier that clearly indicates roles and responsibilities. For example, they are responsible for design controls and managing design changes for the device, while you are responsible for design changes to the labeling. You will also need their support to make sure you are not deviating from the required labeling content that was approved by the regulator (i.e., indications for use, contraindications, warnings, precautions, and instructions). You will need a risk management file that covers labeling, and you will need to maintain a medical device file for the revised labeling.
Since your company is the name on the label, you will be responsible for UDI requirements, complaint handling, adverse event reporting, and recalls.
If your company also modifies the packaging, then packaging validation and distribution testing will also be required. The packaging elements would also be added to your MDF.