The FDA released a new draft guidance document about artificial intelligence and machine learning (AI/ML) functions in medical devices.
What is a predetermined change control plan for artificial intelligence (AI) software?
The new FDA guidance is specific to predetermined change control plans for marketing submissions. The guidance was released on March 30, 2023, but the document is dated April 3, 2023. The draft guidance applies to artificial intelligence (AI) or Machine Learning-Enabled Device Software Functions (ML-DSF), including modifications automatically implemented by the software and modifications to the models implemented manually.
A PCCP must be authorized through 510k, De Novo, or PMA pathways, as appropriate. The purpose of including a PCCP in a marketing submission is to seek premarket authorization for these intended device modifications without necessitating additional marketing submissions for each change described in the PCCP.
How do you determine if a 510k is required for a device modification, and how would a PCCP affect this?
Currently, there are three guidance documents relating to the evaluation of changes and determination if a new premarket submission is required:
These guidance documents will still be the first steps in evaluating changes. Only changes specific to artificial intelligence (AI) or ML-DSF that would result in a new pre-market submission could be subject to a PCCP.
Examples of Employing AI/ML-DSF PCCPs
Retraining a model with more data to improve device performance while maintaining or increasing sensitivity. If this type of change is pre-approved in the PCCP, the labeling can be updated to reflect the improved performance once the change has been implemented.
Extending the scope of compatible hardware with a device system. For example, if the algorithm was initially trained using one specific camera, ultrasound, defined parameter, etc., then a PCCP could add additional cameras/ultrasounds/modified parameters.
Retraining a model to optimize site-specific performance for a specific subset of patients with a particular condition for whom sufficient data was unavailable. The PCCP could expand the indications once such data were available.
What is the difference between a locked vs. adaptive algorithm?
A locked algorithm is a software function involving human input, action, review, and/or decision-making before implementation. Once the algorithm is designed and implemented, it cannot be changed without modifying the source code.
Locked algorithms contrast with adaptive/automatic algorithms, where the software will implement changes without human intervention. The adaptive/automatic algorithms are designed to adjust according to changing input conditions. The adaptive/automatic algorithm is designed to recognize patterns in the input data and adjust its processing accordingly.
Typically locked algorithms apply to fixed functions such as a decision tree, static look-up table, or complex classifier. For AI/ML-DSF, manually implemented algorithms may involve training the algorithm on a new dataset or serving a new function. Once the training is complete, the algorithm will be implemented into the software. Adaptive algorithms are programmed such that their behavior changes over time as it is run based on the information it processes.
As it relates to a PCCP, the detailed description of the intended modifications needs to specify which algorithm type is being modified.
What is included in a PCCP for artificial intelligence (AI) software?
A PCCP should consist of:
Detailed Description of Intended Modifications
Modification Protocol describing the verification and validation activities, including pre-defined acceptance criteria
Impact Assessment identifying the benefits and risks introduced by the changes
The detailed description of the intended modifications should list each proposed device modification and the rationale for each change. If changes require labeling modifications, that should also be described. It should also be clearly stated whether or not the proposed change is intended to be implemented automatically or manually. The description should describe whether the change will be implemented globally across all devices on the market or locally, specific to different devices based on the unique characteristics of the device’s patient or clinical site.
The types of modifications that are appropriate for a PCCP include modifications related to quantitative measurements of ML-DSF performance specifications, changes related to device inputs, and limited modifications relating to the device’s use and performance. The draft guidance provides some examples of each of those modification types.
The content of the modification protocol section requires a description of planned data management practices relating to the reference standard and annotation process, a description of re-training practices and processing steps, performance evaluation methods and acceptance criteria, and internal procedures for implementing updates.
The impact assessment is the documentation of the evaluation of the benefits and risks of implementing the PCCP for the software. Any controls or mitigations of the risks should be described in this section.
Appendix A of the draft guidance includes example elements of modification protocol components for ML-DSFs. Appendix B includes examples of ML-DSF scenarios employing PCCPs.
If, at some point, the manufacturer wants to make changes to the content of the PCCP relating to either the modifications described or the methods used to validate those changes, that generally would require a new marketing submission for the device.
Utilizing a PCCP in your QMS Change Control System
When evaluating and implementing changes, the manufacturer shall do so in accordance with their Quality Management System change control processes. This should require a review of planned modifications against the FDA guidance documents for evaluating changes and the PCCP. For the change to be acceptable under the PCCP, it must be specified in the Description of Modifications and implemented in conformance with the methods and specifications described in the Modification Protocol. A new premarket submission is required if it does not meet those requirements.
Everyone has a favorite resource they use to answer regulatory questions, but can you trust OpenAI or Elsmar to answer correctly?
If you are deathly afraid of trying new technology, the image above is a screen capture from OpenAI describing “itself.” OpenAI is artificial intelligence (AI), but it is not self-aware yet. The image below is a screen capture from the “About” webpage for Elsmar Cove. This article was the oldest post on the Medical Device Academy, and it described how to use Elsmar Cove as a resource for quality systems and regulatory questions. To update that blog, we are comparing the use of OpenAI with Elsmar Cove. Just in case you were wondering, Elsmar Cove is #6 on our list of favorite search tools, and OpenAI is #5:
Search the FDA.gov device databases – 3rd place, you can email DICE or call 301.796.7100
Search Google (world’s #1 search engine) – 4th place, ask any 5-year-old for help
Are the answers provided by OpenAI and Elsmar Cove accurate?
To test the accuracy of a common regulatory question, we chose a question we weren’t 100% sure about when a client asked last month. I asked my team, but nobody was 100% certain. Basil Systems is limited to submission and post-market surveillance data. I searched FDA.gov, but it was not clear. Google gave us a link to the FDA website. I asked a couple of ex-FDA consultants, but they gave me outdated information. On Thursday, March 30, 2023, I asked Lisa King during an AAMI course I was co-teaching. Lisa is a Consumer Safety Officer at the FDA responsible for reviewing device entries into the FDA. She is also in very high demand for public training courses. She said the contract manufacturers used to be exempt from registration if they shipped to a legal manufacturer first. The regulations changed, and now 100% of contract manufacturers making a finished device must register with the FDA. She also clarified that the FDA doesn’t use the term “legal manufacturer.”
As you can see from the above answer provided by OpenAI, the ChatGPT engine [i.e., Model: Default (GPT-3.5)] effectively produces the correct answer. Using the same wording for the regulatory question, “Does a foreign contract manufacturer need to register with the FDA if they are shipping the medical device to the legal manufacturer first before the device is exported to the USA?” there were no results from Elsmar Cove. After several attempts, I found what I was looking for using the following search terms, “FDA registration of contract manufacturers.” There were multiple related search results, but the most useful discussion threads in the Elsmar Cove discussion forum were:
The most succinct correct answer in the forum is copied below.
Can you trust OpenAI and Elsmar Cove to answer your regulatory questions?
OpenAI is only as effective as the data used to train it. This is constantly evolving, but we have identified search results that were 100% accurate, results that were outdated, and results that were scary wrong. The same is true of discussion forums. Elsmar Cove is one of the best discussion forums for the medical device industry, but people also use ASQ, RAPS, and AAMI. The quality of the information provided depends upon the knowledge and experience of the people participating in the forum, but it also depends upon the forum’s moderation. Elsmar Cove has some experienced moderators with decades of experience. There is always the chance that the most experienced person in the world could answer your regulatory question incorrectly. This usually creates a problem because everyone else in the forum hesitates to challenge a recognized expert. Therefore, regardless of which resource(s) you use, always try to get a reference to the trustworthy source of the applicable regulation. Even Lisa King could make a mistake, but she immediately said, you can find the regulations in the US Code of Federal Regulations (i.e., 21 CFR 807). The bottom line is, always do your fact-checking and reference your source(s).
Breaking news! The FDA just released new guidance on the refusal to accept (RTA) policy for cybersecurity devices.
Where can I find the new cybersecurity devices guidance?
The new guidance is titled “Cybersecurity in Medical Devices: Refuse to Accept Policy for Cyber Devices and Related Systems Under Section 524B of the FD&C Act,” and you can download a copy of the PDF directly from our website. This is the first time the FDA has created a definition for a “cyber device,” but this guidance is specific to the refusal to accept policy (RTA) rather than guidance for the format and content of pre-market notification (i.e., 510k) If you want to learn about new guidance documents as they are released, we recommend that you sign up for FDA email notifications. If you want to be notified of when our new blogs are posted, subscribe to our blog email notification list on this page.
What is a “cyber device” in the context of this cybersecurity devices guidance and submissions?
This new guidance defines “cyber device” using the following language:
includes software validated, installed, or authorized by the sponsor as a device or in a device;
has the ability to connect to the internet; and
contains any such technological characteristics validated, installed, or authorized by the sponsor that could be vulnerable to cybersecurity threats.
What does “refusal to accept” (RTA) mean?
“Refusal to accept” or (RTA) is a policy that the FDA implemented for pre-market notification submissions (i.e., 510k) in 2012. The process occurs during the first 15 calendar days of the FDA review process. The FDA assigns a preliminary reviewer to perform the RTA screening of the submission, and the person completes an RTA checklist. The FDA substitutes an RTA screening with a technical screening for FDA eSTAR templates, and this is one of the reasons why Medical Device Academy uses the FDA eSTAR templates for all 510k submissions and De Novo classification requests instead of using the older 510k format and content requirements with 20 sections.
When will the FDA begin rejecting submissions during the RTA processes?
The FDA states directly in the guidance document that they will not reject submissions for cybersecurity for the balance of FY 2023 (i.e., before October 1, 2023). The wording used by the FDA is: “The FDA generally intends not to issue “refuse to accept” (RTA) decisions for premarket submissions for cyber devices that are submitted before October 1, 2023, based solely on information required by section 524B of the FD&C Act. Instead, the FDA will work collaboratively with sponsors of such premarket submissions as part of the interactive and/or deficiency review process.” We believe the FDA will update the eSTAR template to include requirements for cybersecurity on October 1, 2023. It will not be possible to submit a 510k that does not include the cybersecurity requirements in future eSTAR templates, because the eSTAR automatically verifies the completion of each section in the template.
Will there be another cybersecurity guidance released soon?
The FDA announced last October that a new cybersecurity guidance would be replacing the 2014 final guidance for cybersecurity. A draft was released in 2018, and an updated draft was released in 2022. The final updated guidance is included in the A-list of FDA priorities for final guidance documents, but the updated final version has not been released yet. The FDA webpage for cybersecurity was updated to include this new guidance on RTA policy for cybersecurity devices. We believe this indicates that the updated final version will be released soon. When it is released, we will publish a new blog about that guidance.
Would you like to learn nine ways to improve your quality system procedures? One method is precisely the opposite of our advice from 2011.
During a CAPA course I taught on Friday, January 28, 2011, one of the attendees asked if we teach a course on “How to write better quality system procedures.” Unfortunately, we could only offer material from a course about “Training the trainer.” That “Training the trainer” course focused on visual communication. Several books related to Lean Manufacturing explain how to use visual communication to replace text (i.e., “a picture says a thousand words”). During my ride home, however, I thought of a few other ideas that might help anyone writing or re-writing a procedure. The article was updated and posted as a new blog on Tuesday, March 28, 2023.
1. Use a standardized template for your procedures
In 2013 we published a blog about using a procedure template where we described our 12-part procedure template (i.e., TMP-001). You don’t have to mimic our template, but using a template will accelerate the speed of your writing when you create procedures, and it makes sure you don’t forget any of the essential elements. In addition, using templates ensures a consistent format that makes it easier for everyone to find the information they are looking for. Just make sure that your document control procedure allows flexibility to deviate from the template. The ISO 13485:2016 standard does require a “mandatory” format. Referring to your template as “suggested formatting” will avoid unnecessary nonconformities.
2. Create a process “turtle diagram” for each quality procedure
All of the procedures that Medical Device Academy created have a flow chart at the beginning of the procedure showing the procedures and forms associated with processes that are inputs to that procedure and outputs from that procedure. To systematically improve our procedures, we will be systematically replacing those flow charts with turtle diagrams for each process. This will give more detail than our current flow charts, and internal and external auditors can use the turtle diagrams to understand process interactions.
3. Avoid making unnecessary references to regulations and standards
If you are writing a procedure on risk management—it makes sense to reference ISO 14971. It does not make sense to reference all the other risk analysis standards unless you specifically use them to perform risk analysis. ISO 14971:2019, Clause 4.1, also states that you “shall establish, implement, document, and maintain an ongoing process for” risk management activities. However, the ISO 14971 standard is not directly linked to other procedures. Therefore, ISO 14971 should only be referenced in another if you are using it in that procedure or referencing it directly. For example, the Quality Manual (i.e., POL-001) explicitly references ISO 14971. In contrast, the design control procedure (i.e., SYS-008) references the risk management procedure (SYS-010) but doesn’t reference ISO 14971.
Concerning regulations, you should only reference regulations if the procedure meets a specific requirement. Color coding with symbols should demonstrate traceability to requirements (see method #5 below for further explanation). Rather than adding a reference to regulations in a procedure where there is no requirement, a better approach is to indicate in the Quality Manual that only procedures that have specific requirements will reference the regulations, such as 21 CFR 820 or Part 1 of the Canadian MDR.
4. Track standards, regulations, and the version used in your procedures
In the original 2011 version of this article, we advised quality managers to “avoid including the revision of a standard” because “this is just another opportunity for unnecessary nonconformities.” However, we find that our team has trouble identifying every procedure that a change in regulation or a standard might impact. A systematic process is needed to identify every procedure referencing a regulation or standard. Therefore, we will reference all impacted procedures next to the regulation or standard in our Master Document List (i.e., LST-001). References to the regulations will be added to the main tab for policies, procedures, and work instructions (i.e., [POL, SYS, and WI]). References to the standards will be added to the tab for documents of external origin (i.e., [Doc Ext Origin]).
Many people feel that you should not reference the version of a standard in a procedure because adding the version of the standard increases the number of documents that need to be updated when a standard changes. However, if you are only referencing standards in procedures when it is necessary, then that procedure should be reviewed and updated for the need to be changed. Updating the version of the Standard referenced is the best way to document that a gap analysis against the new version has been completed and the necessary updates were made to the procedure.
5. Use color coding and symbols in your quality system procedures
Matthew Walker, Medical Device Academy’s manager of the human factors team, has systematically updated many of our procedures to the EU Medical Device Regulations 2017/745 and the In Vitro Diagnostic Regulations 2017/746. When he updates our procedures, he references the regulations and applicable ISO 13485:2016 clauses. During certification audits, certification body auditors sometimes have difficulty finding where specific requirements are located in the procedures. Therefore, Matthew added color-coded clause references for our clients and auditors as a corrective action. To make the procedures inclusive for people that are color-blind, Matthew added symbols to supplement the color coding. The extra addition of symbols has proven invaluable because now anyone can search the documents electronically for a symbol to find where all the references are located.
6. Indicate the process owner and training requirements associated with each procedure
Identifying the process owner and training requirements in every procedure makes it easier to define who is responsible for reviewing and revising procedures. For the training requirements, the process owner should specify who needs to be trained on the process. Why? They know the procedure best. If there is a “grey area,” this should be resolved with the department manager for the job function. In addition, retraining requirements should be specified. The training section should also clarify if retraining is required when revising a procedure. If the revision is minor, training should only be necessary for people not trained on a previous revision.
7. Adopt thePlan-Do-Check-Act (PDCA) model for the structure of quality system procedures
For the “Plan” portion, the procedure should explain how to prepare to do something. This planning activity can apply to anything from planning to perform an audit to planning to inspect incoming raw materials. The “Do” portion is what most people refer to as the “Procedure” section. The “Check” portion of the procedure is a great place to specify the monitoring and measurement requirements for the process (see Section 8.1 of the Standard). Finally, the “Act” portion of the procedure should indicate what to do when target metrics are unmet. For example, what should be done when an alert limit is reached? What should be done when an action limit is reached?
8. Include therevision history of quality system procedures
It’s helpful to know which Document Change Notice (DCN) approved the document revision, why the changes were made, the nature of the changes, whether there is a related corrective action, and when the change was made. This will also tell auditors whether there is anything new to audit since the previous internal or external audit. This section is usually near the beginning of our procedures, but it doesn’t matter if the revision history is at the end or the beginning. However, it does help to be consistent.
9. Identify the form number, location, and retention period for each record
We have a section about quality system records near the end of every procedure. This section lists each quality system record that is associated with the procedure. The relevant form is referenced, but we recommend storing these records in electronic or paper folders labeled with the form number. If the files are digital, a hyperlink should be included. If the files are paper, then you should list the physical location of storage. The retention period can be listed in each procedure. Still, it will be essential to ensure that this information matches the regulatory requirements and record retention requirements in your “Control of Records” procedure (i.e., SYS-002).
This article gives you five ways a management representative can demonstrate value to medical device top management teams.
Align quality objectives with the company first and the FDA second
A fast way to alienate yourself as a management representative is to begin every conversation with a quote from the FDA regulations. Instead, ensure that quality objectives align with the company’s overall goals. For example:
Complete the design verification and validation of our new product by August 15.
Reduce nonconforming products from the molding process by 50% this year.
Increase the number of production lots released each week from four to five lots of 1,000 units per lot.
Next, ensure that your quality objectives are achievable, measurable, and have clear timelines for completion. Quality objectives should not be stretch goals. If you have to initiate a corrective actionbecause you didn’t achieve a quality objective, you just create more work for yourself and the company.
Teach people to focus on the process and not the procedure
The FDA and the ISO 13485 standard require procedures to be established. However, if you focus on the documentation of processes, your company will do stupid things faster. Instead, management representatives need to be able to teach people how to make processes more effective before the processes are documented. Lean manufacturing techniques are not limited to manufacturing. You can apply lean methods to administrative processes too. For example:
What information needs to be in a form?
What is the correct order of tasks for the process?
Is there duplicate or unnecessary information?
A management representative helps identify what to measure
In a management review meeting, the effectiveness of the quality system is reviewed, and improvements are identified. This does not mean the management representative needs to measure or create slides and graphs. As a management representative, you should ask the CEO the most important information they want from each department or member of top management. Once you know what information the CEO wants, please work with the other members of top management to find the most efficient way to get that information and graph it. Help the other managers identify who can generate the graph with the least effort (it’s seldom a manager), and help that person build the reporting of that information into their routine.
A management representative needs to share the spotlight
A management review meeting is only effective if the top management is engaged in the process. Therefore, the management representative should not create 100% of the slides or present 100% of the slides. Everyone should have a piece they are responsible for and can be proud of. When an individual or a team achieves a goal, we can celebrate the achievement in a management review. When an individual or team struggles, we can ask for help in a management review. If other members of top management are not engaged in preparation for a management review, they will not be enthusiastic about listening to the presentation either.
Have a positive attitude as a management representative
Everyone hates to listen to someone that has a negative attitude. As managers, we sometimes need to report bad news. However, we need to develop ideas to solve problems instead of just reporting gloom and doom. We also need to ensure we never miss an opportunity to report good news.
Management representatives should schedule reviews more often
This last section is a bonus (i.e., a sixth way to ensure you are a valuable management representative). Most management review procedures require a management review at least once per year. Unfortunately, there is little point in reviewing quality information from last February during this January. If changes to your quality system are planned or implemented, more frequent reviews are needed. Examples of changes that should prompt you to schedule an extra management review include mergers, new product launches, and employee turnover.
The FDA eSTAR includes a list of eight different options for a sterilization method, but how do you select the best method and validate it?
What is Sterile Packaging Day?
The Sterilization Packaging Manufacturers Council (SPMC) founded Sterile Packaging Day in 2021 to recognize and thank all of the companies in the supply chain who work together to deliver innovative, safe, and sterilized devices to provide excellence in patient care. Sterile Packaging Day is February 8, 2023, and this year’s celebration theme is “Designed to Protect.” SPMC provides four tips for celebrating Sterile Packaging Day:
Tell us in one word what “Designed to Protect” means to you (Rob chose “Lifesaving”)
Thank you to Jan Gates!
How to select the best sterilization method
Several factors determine the best sterilization method to use for your device. The first factor is whether your device will be delivered sterile or will the end user sterilize the device. If the end user is responsible for sterilizing the device, the most common methods used by hospitals are:
steam sterilization
hydrogen peroxide sterilization
EO sterilization
The popularity of the third method is declining due to environmental restrictions on hazardous emissions from the ethylene oxide sterilization process. Hydrogen peroxide is gaining popularity because it can be used for heat-sensitive materials, and hydrogen peroxide vapor reacts with moisture to form a harmless aqueous solution. Steam is the most common sterilization method used by doctors, dentists, and hospitals because steam sterilizers are relatively inexpensive, and no hazardous chemicals are required.
The second factor to consider when selecting a sterilization method is whether there are any heat-sensitive components. Plastics will melt and degrade in dry heat sterilization cycles, and some plastics cannot withstand the temperature of a steam sterilizer. Therefore, if your device is constructed from plastics for cost reduction, weight, magnetic resonance (MR) compatibility, or other reasons, you may need to use a sterilization method with a lower temperature process.
The third factor to consider when selecting a sterilization method is whether any long, narrow tubes require sterilization. These design features are difficult to sterilize for any vapor-based sterilization process, such as steam, hydrogen peroxide, or ethylene oxide. There are a few process control strategies that can be used to sterilize with gas:
use of an extreme vacuum to improve penetration of sterilant gas
ensuring that the device and packaging materials are dry
use of longer cycles with more sterilant gas
use of internal biological indicators at the most difficult sterilization location
The fourth factor to consider when selecting a sterilization method is whether the device includes a liquid. A liquid cannot be sterilized with hydrogen peroxide, ethylene oxide, or dry heat. In some cases, the liquid may be a sterilant (i.e., ISO 14160:2021 for liquid chemical sterilizing agents). There are three popular solutions for the sterilization of a device that includes liquid:
steam sterilization–assuming the liquid doesn’t contain components that are heat sensitive (e.g., proteins)
filter sterilization–usually combined with aseptic filling and pre-sterilizing containers)
radiation sterilization with eBeam or Gamma
eBeam and Gamma are also used for sterilizing products where cross-linkage of ultra-high molecular weight polyethylene (UHMWPE) is desired, or it is impossible for a gas sterilant to penetrate all areas of a device.
What are the applicable sterilization validation standards for each sterilization method?
As shown in the FDA eSTAR screen capture above, eight possible sterilization methods can be selected for sterilizing a medical device in a 510k or De Novo submission. Each sterilization method has a different applicable standard that should be used to validate the sterilization process, but in all cases, the sterilization process must result in a sterility assurance level (SAL) of 10-6.
The FDA feels that the Established A (Est A) methods of sterilization have a long history of safe and effective use, while the FDA has not recognized a dedicated consensus standard for the Established B (Est B) sterilization methods. However, there are examples of devices that have received FDA 510k clearance using each of the non-traditional sterilization methods (i.e., Est B methods). Manufacturers will generally adapt existing international standards for sterilization validation to validate the non-traditional methods. There is published information on the development, validation, and routine control for these non-traditional sterilization processes.
Links to each of the recognized standards are provided below:
Steam (Moist Heat) (Est A) – ISO 17665-1:2006, Sterilization of health care products — Moist heat — Part 1: Requirements for the development, validation, and routine control of a sterilization process for medical devices
Ethylene Oxide (EO, EtO) (Est A) – ISO 11135:2014, Sterilization of health care products – Ethylene oxide – Requirements for development, validation and routine control of a sterilization process for medical devices; and ISO 10993-7:2008, Biological evaluation of medical devices – Part 7: Ethylene oxide sterilization residuals
Radiation (Est A) – ISO 11137-1:2006, Sterilization of health care products – Radiation – Part 1: Requirements for development, validation, and routine control of a sterilization process for medical devices; ISO 11137-2:2013, Sterilization of health care products – Radiation – Part 2: Establishing the sterilization dose
Dry Heat (Est A) – ISO 20857:2010, Sterilization of health care products – Dry heat – Requirements for the development, validation and routine control of a sterilization process for medical devices
Hydrogen Peroxide (Est B) – ISO 22441:2022, Sterilization of health care products — Low temperature vaporized hydrogen peroxide — Requirements for the development, validation and routine control of a sterilization process for medical devices (this standard is not recognized by the US FDA)
Ozone (Est B) – this is a new method using Ozone gas, and the method of action is similar to EO and H2O2
Flexible Bag Systems (Est B) – ISO 22441:2022 should be used for validation of flexible bag systems with hydrogen peroxide, but instead of validating the process with three half-cycles that are half the duration of the full-cycle, instead, you use three half-cycles that use half the volume of sterilant of a full-cycle; this method is used by Andersen Scientific for their EO Bag sterilizers.
Novel Methods – ISO 14937:2009, Sterilization of health care products – General requirements for characterization of a sterilizing agent and the development, validation and routine control of a sterilization process for medical devices
When should you use a novel sterilization method?
Novel sterilization methods should only be used when none of the traditional (Est A) and non-traditional (Est B) sterilization methods will not work. For example, aseptic filling combined with filtration of liquids is a common strategy for pre-filled syringes if the liquid is sensitive to radiation sterilization. Sterilization with peracetic acid has been used for a long time, but the sterilization method has not gained widespread popularity. Peracetic acid can also be combined with hydrogen peroxide. There is also a low-temperature steam and formaldehyde validation standard (i.e., ISO 25424:2019). Sterilization with UV light is a process that is sometimes used where materials are sensitive to high temperatures and where all surfaces can be penetrated with UV light. Nitrogen dioxide was developed as a more environmentally friendly sterilant similar to ethylene oxide. X-Ray is a new type of radiation sterilization that is being developed as a high-speed alternative to Gamma and eBeam, but X-Ray sterilization also has the advantage of being able to control a narrower dose range than Gamma and eBeam processes.
Consensus Standards for Sterilization Validation
There are also additional supporting standards that you will need for validation of your sterilization process. The following is a partial list of the standards you might consider:
ISO 11737-1:2018, Bioburden Testing for Aerobic Bacteria and Fungi
USP<51> Antimicrobial Effectiveness Test
Candida albicans (a yeast…yeasts are a form of fungus)
Aspergillus brasiliensis (a filamentous mold…also a fungus)
Escherichia coli (a bacterium…better known as “E. coli”)
Pseudomonas aeruginosa (a bacterium….very problematic industrially)
Staphylococcus aureus (a bacterium…better known as “Staph”
USP<61> Bioburden or Microbial Limits Test (Total Aerobic Microbial Count = TAMC; Total Yeast and Mold Count = TYMC)
USP<62> Objectionable Organisms or Pathogens Tests
ISO 11138-1:2017, Sterilization of health care products – Biological Indicators – Part 1: General Requirements
ISO 111140-5:2017, Sterilization of health care products – Chemical indicators – Part 5: Class 2 indicators for Bowie and Dick air removal test sheets and packs
ISO 17664-1:2021, Processing of health care products – Information to be provided by the medical device manufacturer for the processing of medical devices – Part 1: Critical and semi-critical medical devices
Aging and Shelf-life Testing
The current standard for accelerated aging studies is ASTM F1980:2021 “Standard Guide for Accelerated Aging of Sterile Barrier Systems and Medical Devices has been revised and recently released to include medical devices.” Jan Gates explains that the “and” used to say “for.” The language was updated with more information on product humidity effects to go with the title. Jan was kind enough to write a Shelf-life Testing Protocol for us based on this new version of the standard. The protocol includes requirements for real-time and accelerated age testing of a product. If you need basic training on how to validate the shelf-life of your device, we have a webinar for sale on sterility and shelf-life. We also recorded an updated webinar on January 19, 2023, as part of the FDA eSTAR updates to our 510(k) Course.
Distribution Conditioning Tests & Packaging Performance Tests
Where can you find a procedure for each sterilization method?
ISO 13485:2016, Clause 7.5.7 is specific to the “Particular requirements for validation of processes for sterilization and sterile barrier systems.” This clause includes the requirement to establish procedures for sterilization validation and validation of your sterile barrier systems. Even if your company uses a protocol and procedures established by a contract manufacturer, you still need to establish an internal procedure(s) to meet this requirement if you have sterile products. The following is a list of procedures sold by Medical Device Academy:
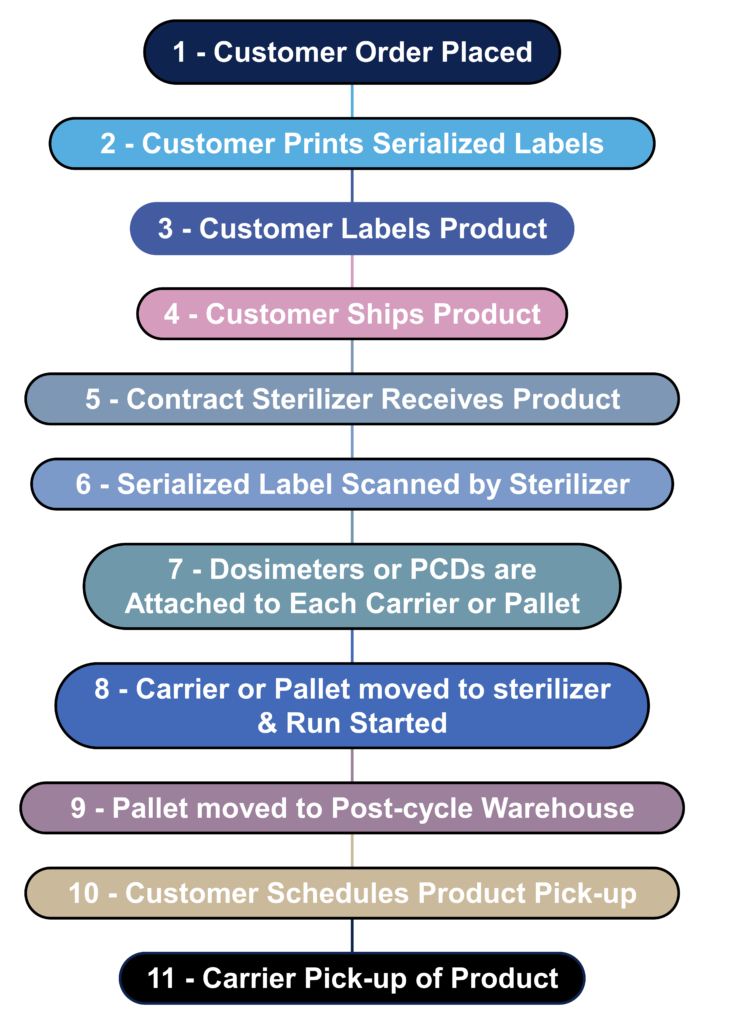
What is the process flow for contract sterilization?
Most device manufacturers do not sterilize their devices in-house. Instead, sterilization is outsourced to a contract sterilizer. The process flow diagram below is a hypothetical process flow diagram for a contract sterilization process. The only step not included in this process flow is the incubation of biological indicators because gamma and eBeam sterilization processes use dosimeters instead of biological indicators. The nature of biological indicators is also changing rapidly because manufacturers are developing “rapid test” biological indicators. In 2008 I worked extensively with self-contained biological indicators that eliminated the need to use an aseptic technique to transfer biological indicators into culture media. In addition, I complete an incubation reduction study to validate a shorter 48-hour incubation cycle instead of the typical 7-day sterility test. Terragene is one of the manufacturers developing next-generation technology for biological indicators that allows the results to be read within seconds instead of 48 hours. This next-generation technology also incorporates barcode readers and networked readers to ensure traceability to each biological indicator and reader.
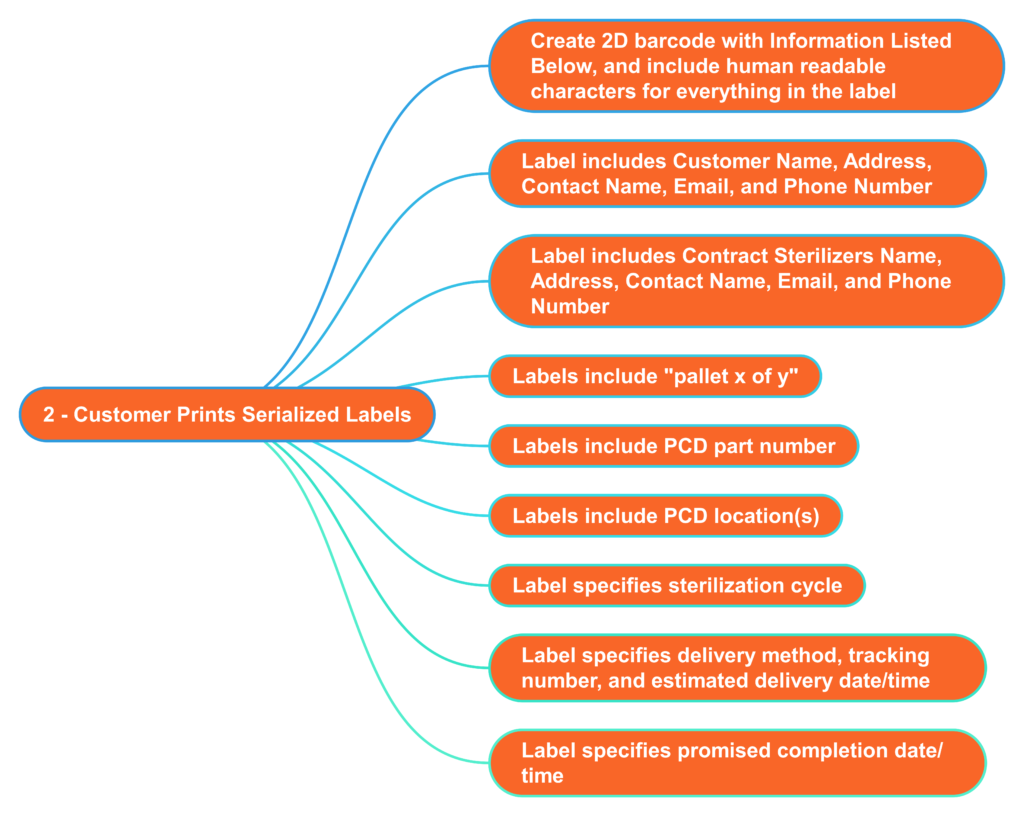
What information should serialized labels include for contract sterilizers?
In the “olden days” (c. 2005), I used to print out labels for each pallet that we shipped to the Isomedix facility in Northboro, MA. The label identified who the product was from and what we wanted Isomedix to do with the product (e.g., gamma sterilize at 25-40 kGy). At the time, we were just beginning to incorporate barcodes into on-demand labeling to facilitate traceability. 18 years later, companies are still stalling the implementation of on-demand barcoded labels. Almost every shipping dock has a barcode reader, and the technology is inexpensive. Therefore, you should consider creating a template for on-demand barcoded labels with all the information listed below. This will reduce the risk of errors by the contract sterilizer and enable you to identify when a mistake was made quickly. Contract sterilizers should also demand this information on product labeling as an added risk control. All biological indicators and dosimeters are labeled with UDI barcodes now. Therefore, contract sterilizers should be able to create robust process controls that ensure traceability between barcodes on your labeled product with barcodes on the biological indicator or dosimeter.
Read this article to learn why ISO 19011 standard is a vital guidance for anyone that audits quality systems or manages an audit program.
What is ISO 19011?
ISO 19011 is a seven-part international standard for auditing management systems. The standard defines the eight principles of auditing (e.g., the process approach to auditing), provides guidance on managing audit programs and conducting audits, and includes recommendations for evaluating people for competency. There is also an appendix with details on conducting on-site and remote audits.
If you have ever taken a lead auditor course forISO 13485, or one of the other quality management system standards, one of the critical handouts for the class should have been ISO 19011. The title is “Guidelines for Auditing Quality Management Systems.” In 2018, ISO 19011 was updated, and the changes were not superficial. If you need to purchase a copy of ISO 19011:2018, the Estonian Center for Standardization and Accreditation is the least expensive source we know.
ISO 19011 covers the topic of quality management system auditing. This Standard provides guidance on managing audit programs, conducting internal and external audits, and determining auditor competency. One of the most common points of confusion in the lead auditor course is the difference between first, second, and third-party audits. In the first edition of this Standard, the difference between first, second, and third-party audits was just a note at the bottom of page one and the top of page two. The note was also not clear. In the second edition of 19011, in Table 1 (reproduced below), the difference between these three types of auditing is crystal clear. Table 1 was modified further in the 3rd edition to include a bottom row that remains unchanged in the 3rd edition, released in 2018.
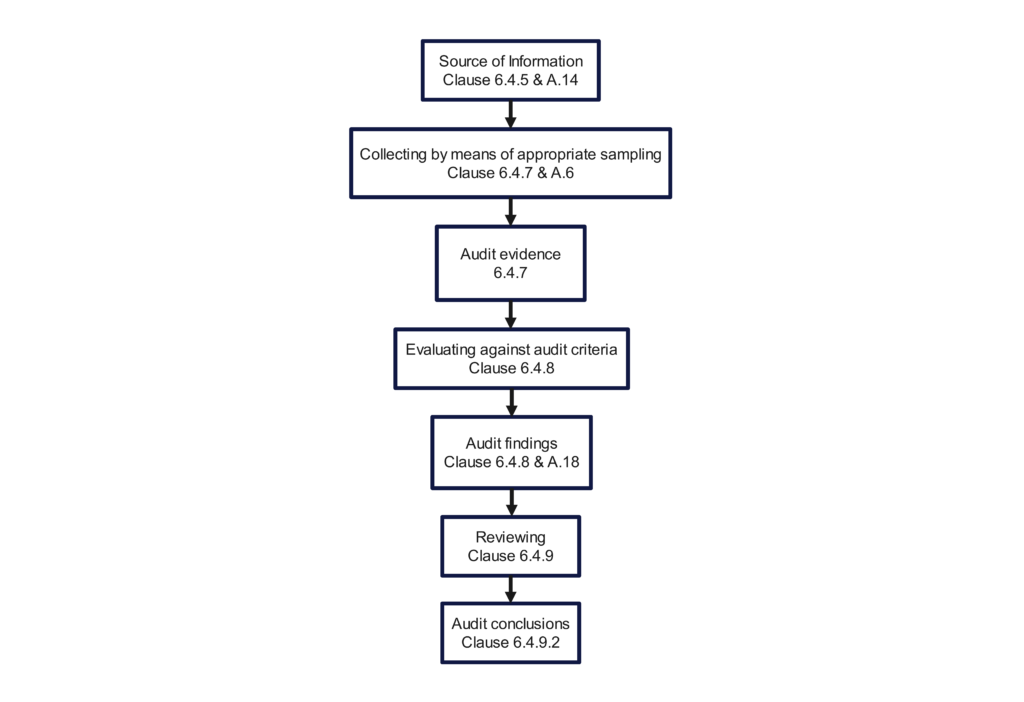
Figure 1, found in Clause 5.1 of the 2nd edition, was combined with Figure 2, found in Clause 6.1 of the 2nd edition. The combined figure is now Figure 1 in the 3rd edition. The combined scope of Figure 1 is now a “Process flow for the management of an audit program” and a “Process flow for conducting an audit.” The figure categorizes the various stages of audit program management and conducting an audit into the Plan-Do-Check-Act (PDCA) cycle. We highly recommend this style for presenting any process in your internal procedures as an example of best practices in writing an SOP. The flow chart even references each of the clauses in the Standard.
The 2018 version still includes an opening meeting checklist (i.e., Clause 6.4.3) and a closing meeting checklist (i.e., Clause 6.4.10). Figure 3 in the 2nd edition, “Overview of the process of collecting and verifying information,” was a poor example of a flow chart. The committee did not update the figure when the standard was updated for the 3rd edition. Therefore, we updated the figure below to provide additional traceability to the Clauses of the Standard. If you incorporate this figure into your quality auditing procedure, you should substitute references to your procedure’s sections instead of the clauses of the standard.
Competency Requirements in ISO 19011
Many audit procedures neglect to define the qualifications and methods for determining thecompetency of the audit program manager. Clause 5.3.2 tells you how. Put it in your own procedure. Most of the procedures we read include qualifications for a “Lead Auditor,” but we seldom see anything regarding competency. Unfortunately, this Standard only explicitly addresses the “Lead Auditor” competency in a two-sentence paragraph—Clause 7.2.5. When we teach people how to be Lead Auditors, we spend more than an hour on this topic alone.
The Standard would be more effective by providing an example of how third-party auditors become qualified as a Lead Auditor. Third-party accreditation requires the auditor to be an “acting lead” for audit preparation, opening meetings, conducting the audit, closing meetings, and final preparation/distribution of the audit report. This must be performed for 15 certification audits (i.e., – Stage 2 certification or re-certification), and another qualified lead auditor must evaluate you and provide feedback.
Appendices in ISO 19011
The appendices were the last significant additions to this Standard in 2011 (i.e., 2nd edition). Annex A provided examples of discipline-specific knowledge and skills of auditors. This section was eliminated from the 3rd edition of ISO 19011:
“Due to the large number of individual management system standards, it would not be practical to include competence requirements for all disciplines.” – Copied from the Foreward
I think providing adding a short Annex to each management system standard that defines recommended discipline-specific knowledge would be helpful. Still, that kind of change would need to be initiated with the next version of ISO 9001.
Appendix B in the 2nd edition is now Appendix A in the 3rd edition of ISO 19011. A table (Table A.1 – Audit Methods) compares conducting on-site and remote audits. We were pleased to see that conducting interviews is a significant part of remote auditing in this table. Section A.17 in the appendix provides suggestions for conducting interviews. Still, if you exhibit all 13 professional behavior traits found in Clause 7.2.2, you don’t need advice on speaking with people. For the rest of us mortals, we could use a five-day course on interviewing alone. To improve your skills in this area, ask an experienced auditor with solid interviewing skills to watch and comment on a recording of a virtual audit you perform. Watching yourself audit is cringe-worthy, but we guarantee you will improve.
What are the primary changes to the 2018 version of the standard?
There are seven main differences between the second edition, published in 2011, and the third edition of ISO 19011, released in 2018:
addition of a seventh principle of auditing in sub-clause 4(g) (i.e., risk-based approach);
more guidance on audit program management in Clause 5, including audit program risk;
expansion of Clause 6 on conducting an audit–especially Clause 6.3 on audit planning;
expansion of auditor competence requirements in Clause 7;
updating of terminology to emphasize processes rather than objects;
removal of an annex containing competence requirements for specific quality management systems;
expansion of Annex A to include guidance on new auditing concepts such as remote audits.
Risk-based auditing is the most significant change in the 2018 version of ISO 19011
One of the main differences between ISO 19011:2018 and the previous 2011 version is the addition of a “risk-based approach” to the principles of auditing. Specifically, clause 4(g) of the guidelines for auditing management systems is, “The risk-based approach should substantively influence the planning, conducting and reporting of audits to ensure that audits are focused on matters that are significant for the audit client, and for achieving the audit program objectives.” A lot of people are unsure of what is meant by a risk-based approach. Still, the key to understanding this is to focus on the definition of risk. From a product perspective, the risk is the “combination of the probability of occurrence of harm and the severity of that harm.” From a process perspective, the risk is the “effect of uncertainty on an expected result” (ISO 9001:2015, clause 3.09). Therefore, auditors should emphasize medical devices with the highest severity of harm and devices with a high probability of hazards or hazardous situations. When an auditor focuses on a process rather than a specific medical device, auditors should emphasize any processes that are not under control and any recent process changes.
What is risk-based auditing?
Risk-based auditing considers the risks of failing to achieve audit objectives and the opportunities created by choosing various audit methods and strategies. For example, a desktop audit of procedures might be appropriate if you are conducting your first internal audit for a new quality system. Alternatively, a desktop audit would be a waste of time if you are auditing a mature quality system where very few changes to procedures have been made in the past year. Using the element approach to auditing is unlikely to add much value. Audits are meant to be a sampling. Therefore, you should focus on areas of importance where previous nonconformities were identified, any new products or processes, and anything that changed significantly.
Auditor selection should also be risk-based
Suppose you are conducting a supplier audit as part of your initial supplier qualification for a critical component supplier or contract manufacturer. In that case, you should consider doing a team audit with a multi-disciplinary team. This is a risk-based approach to the supplier qualification process, which ensures that subject matter experts evaluate each process instead of auditors with a general quality assurance background. This approach also forces more of your personnel to introduce themselves to the new supplier, and the audit will develop more reliable communication channels between your two companies. Alternatively, if you are conducting a routine internal audit of a production process, you might select a new lead auditor to conduct the audit. You don’t expect any significant findings in a routine internal audit of an established production process. In your role as an audit program manager, you need to match the new lead auditor to a process that will force them to look at all aspects of the process approach to auditing. Specifically, process validation, calibration, maintenance, and process monitoring may not apply to other administrative process areas, such as purchasing.
Risk-based auditing should influence your auditing schedule
The frequency of auditing suppliers and internal process areas should reflect the associated risks. Therefore, when you create or update your auditing schedule, you should consider the risk level of the products being audited and the process being audited. Production processes with a moderate or high level of non-conforming products may need to be audited more than once yearly. Still, a supplier with an excellent track record of extremely high quality and on-time delivery may be audited in alternating years. If you previously scheduled a remote audit, you may want to alternate to conducting an on-site audit the next time.
The duration of your audits should not always be the same either. Suppose one production process makes one product in low volume, and another production process makes multiple products in high volume. In that case, you should not schedule a two-hour internal audit for both processes every year. The low-volume production process may only need a one-hour audit once per year. In contrast, the high-volume process may require a four-hour internal audit or multiple annual audits.
Risk-based auditing applied to remote supplier auditing
The risk-based auditing approach was added to ISO 19011:2018 as the seventh principle of auditing. This represents the most significant change to that standard, but how does it apply to remote auditing? Despite the opportunities created by remote auditing, there are also risks associated with auditing suppliers remotely. People worry about auditees hiding hazardous situations or unacceptable environmental conditions such as filth or disrepair. However, unacceptable cleanliness and maintenance practices don’t happen overnight. Therefore, you should expect a clean and well-maintained facility to remain that way. One approach is to alternate between remote and on-site audits to verify the overall condition of a supplier’s facility. Therefore, the risk of auditees hiding objective evidence is more an issue of trust than a highly probable occurrence.
The more probable risks associated with remote auditing are related to the potential lack of availability of records. This is especially important for paper-based quality systems. Most people try to address this risk by scanning paper documents and records, but scanning documents have limited value. Scanning paper documents is more efficiently performed in a large batch by an automated or semi-automated process. Also, auditors and inspectors typically focus on the most recent records, and auditors and inspectors rarely sample 100% of the records. Therefore, the best risk controls include the following:
Ask a guide to send a digital picture of the record.
Use a tripod-mounted HD webcam focused on a music stand or similar surface.
Ask the auditee to read the document while you take notes.
In our experience, you will probably rely on all three risk controls, but it is unlikely to delay the audit. However, in response to the limited physical access to medical device facilities and personnel, certification bodies are sending out questionnaires to assess the risk of being unable to achieve audit objectives or cover the required scope of surveillance and recertification audits. As the audit program manager, you can reduce these risks by working with supply chain managers to develop new supplier questionnaires that specifically ask questions about the capability of supporting audits remotely. In particular, it would be essential to obtain facility maps to identify areas with inadequate cellular coverage and identify records that are only available in hardcopy format.
The 2022 draft cybersecurity guidance from the FDA emphasizes the need to design devices to be secure and the need to design devices capable of reducing emerging cybersecurity risks throughout the total product lifecycle. Designing devices for security must be built into your original design plan, or you will need to modify your device for improved security just to obtain initial 510(k) clearance from the FDA. What is not clear from the guidance or standards is when you need to conduct security testing or repeat tests.
Planning Cybersecurity Tests
As with all quality system processes, cybersecurity testing should begin with a plan. There are two models typically used for the design and development process: Waterfall Diagram (typical of hardware development) and V-Diagram (typical of software development).
Waterfall Diagram
V-Diagram
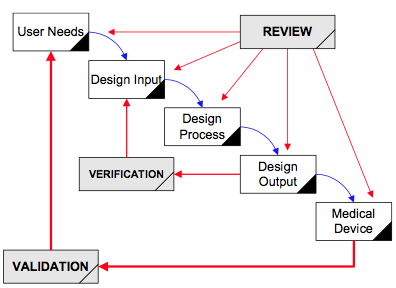
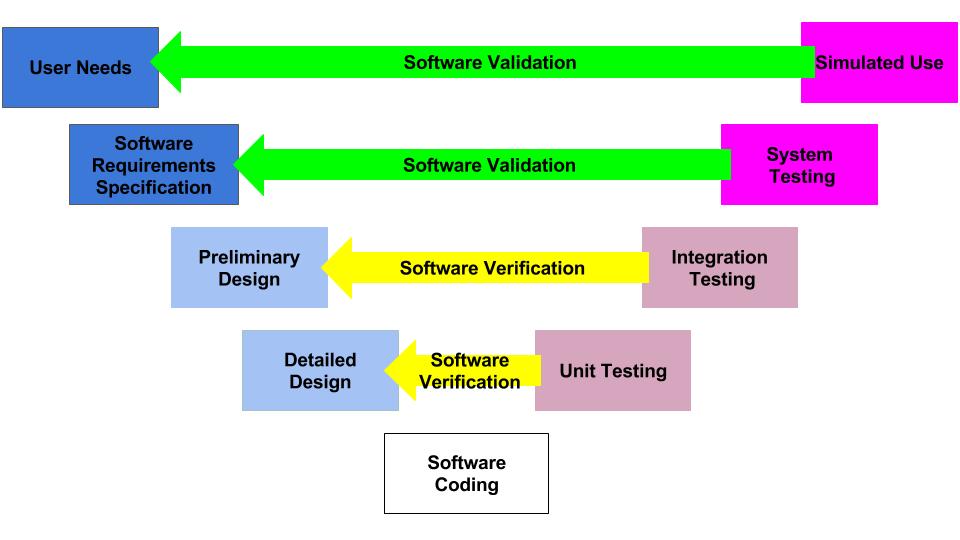
How are design plans for SaMD different from other design plans?
Most of the verification testing for software as a medical device (SaMD) is 1) conducted virtually, 2) tests software code in a “sandbox,” and 3) involves internally developed testing protocols. In contrast, verification testing for other types of devices involves 1) physical devices, 2) testing at a 3rd party lab, and 3) involves international standards and testing methods. The biggest differences between SaMD verification testing and other device verification testing are the speed and cost of the testing. SaMD verification is much faster and less expensive. Therefore, if your software design documentation is efficient, you can complete more design iterations. This is why software developers use the V-diagram to model the design and development process instead of the “waterfall” diagram.
Where do the requirements to test cybersecurity belong in your design plan?
A design plan documents the design and development process for your device. You must establish, maintain, and update the plan as the project progresses. There is no required format, but auditors and the FDA will audit your Design History File (DHF) for compliance with your plan. You are required to document the following content in your plan:
Stages of development
Reviews at each design and development stage
Verification, validation, and design transfer activities at each stage
Responsibilities and authorities for the design project
Methods you are using to ensure traceability of user needs, software hazards, software requirements, software design specifications, and software testing reports
Human resources needed for your design project, including competency
Software Design Inputs
In the early stages of the software development lifecycle, you must select an appropriate threat model and perform a hazard analysis for software security. These security hazards need to be included as design inputs in your software requirements specification (SRS). The need for updateability and patchability should also be included as design inputs.
In parallel with your SRS, you will need to create a User Specification. The SRS and User Specification will determine the use cases and call-flow views that require verification testing later in your software development process. After the SRS has been approved, you will need to create a software design specification (SDS). Each item in the SDS should be traceable to an item in the SRS. The SDS items that trace to security hazards are your risk controls. Each risk control will require you to test cybersecurity to verify risk control effectiveness. At this point, you will need to create your testing protocols for security.
System Testing Protocols to Test Cybersecurity
Testing protocols should include a boundary analysis and rationale for boundary assumptions. Testing protocols should also include vulnerability testing. The FDA recommends the following vulnerability testing:
Abuse cases, malformed, and unexpected inputs,
Robustness
Fuzz testing
Attack surface analysis,
Vulnerability chaining,
Closed box testing of known vulnerability scanning,
Software composition analysis of binary executable files, and
Static and dynamic code analysis, including testing for credentials that are “hardcoded,” default, easily guessed, and easily compromised.
Does your development budget include security testing?
Design control training traditionally emphasizes the importance of “freezing” design outputs before starting verification testing to prevent the need for repeating any of the verification testing. The reason for this is that verification testing is expensive, and it is time-consuming to produce additional verification samples. In contrast, SaMD is guaranteed to be changed multiple times during the verification testing process as software bugs are identified. Therefore, software developers focus on the velocity of developing code and testing that code. One exception to this is penetration testing. Penetration testing is usually conducted once your code is final because it is more expensive than other software verification and validation testing and it would need to be repeated each time the software is updated or patched.
Penetration Testing
Penetration testing is another method used to test cybersecurity that would probably be conducted in parallel with simulated use testing to validate performance and the effectiveness of human factors risk controls. Penetration testing could be at the system level in a sandbox environment, or it can be performed on a sample device in a simulated use environment. Your penetration testing documentation should include the following:
independence and technical expertise
scope of testing
duration of testing
testing employed, and
test results, findings, and observations
Postmarket cybersecurity management
For CE Marked products, there is a requirement for a postmarket surveillance plan (i.e., PMS plan) to be submitted as part of your technical file. The US FDA does not currently have this requirement for Class 1 and Class 2 devices, but Class 3 devices (i.e., PMA) and devices with humanitarian device exemptions (HDE) are required to submit a PMS plan as part of the premarket submission. The US FDA also requires a postmarket cybersecurity management plan to be submitted for premarket submissions of Class 2 and Class 3 devices. You should create your postmarket cybersecurity management plan during your verification and validation activities, and the final version should be approved at the time of product release.
If you need additional resources or training related to cybersecurity, you may be interested in the following:
This blog revies some practical and effective management skills that all managers should possess.
Sometimes we hear phrases like: “Well, that’s just an ISO requirement.” This apparent lack of support by top management is what frustrates every Management Representative in the world.
Peer Support
For a Quality Manager or any manager, it is vital to gain support from our peers, as failure to do so can lead to challenges. While the Quality Department plays a crucial role in recommending improvements, providing training, and assisting with implementation, it cannot address all problems on its own. Therefore, I strongly believe in assigning corrective actions to the process owner (i.e., the Manager) responsible for the area where the problems originated. This approach creates an opportunity for QA/RA to collaborate with the area manager and work together as a team towards the shared goal of improvement.
Good managers build people up and improve processes, they don’t point fingers or blame individuals. It is the process, not the person.
Example of ‘not-an-effective-manager’
Persuading Skeptics
If you encounter resistance when trying to persuade skeptics, focus on a crucial project for the individual opposing your ideas. Demonstrate how applying Quality principles can effectively resolve their problems, potentially gaining their support. Converting one person often leads to strong support from them. If the resistant individual holds a senior position such as the CEO, take time to understand the CEO’s initiatives (These shouldn’t be hard to identify as they likely talk about them rather constantly). Illustrate how their actions can align with Quality Objectives, using graphs and presenting well-thought-out solutions to their challenges. Utilize the CAPA (Corrective and Preventive Action) process as a framework to show how the management team can collaboratively address issues.
If nothing seems to be working, you can always try reviewing some FDA MedWatch reports too–just to scare your boss.
Here is a list of tips to deal with unsupportive top management in a quality management system using effective management skills:
Clearly communicate the benefits of the quality management system:
Articulate the advantages that a well-implemented quality management system can bring to the organization, such as improved efficiency, reduced costs, and enhanced customer satisfaction. Don’t just leave the conversation at “The QSR/13485 says that we SHALL have one”.
Address specific concerns and show how quality initiatives overcome challenges:
Listen to the concerns of top management and present how quality initiatives directly address those issues, fostering a more positive outlook towards the system. Just like the old saying,
“An ounce of prevention is worth more than a pound of the cure”
Consider how ISO 13485:2016 has separate sub-clauses for Corrective Actions and Preventive Actions. Explain how something like pushing for preventive actions shows compliance with clause 8.5.3. which auditors, and inspectors will be looking for, but also that every Preventive Action represents a dodged 483 letter or recall.
Or how beefing up incoming inspection is likely to save time and money in reworked product and less scrap dispositions because any non-conforming materials are stopped before they can make their way into finished devices.
Demonstrate how quality aligns with overall business objectives:
Connect the quality management system to the organization’s strategic goals, highlighting how it contributes to long-term success and profitability. Reframe the Quality Policy and Quality Objectives as tools to support a successful business. Not just, “We have to have them for compliance….”
Start with small projects and showcase measurable results:
Begin with pilot projects or smaller initiatives that demonstrate tangible improvements, instilling confidence and support from top management.
Create a compelling business case for the quality management system:
Develop a well-researched and data-driven business case that outlines the return on investment and the long-term benefits of implementing the system. Effective management skills will involve encorporating topics like regulatory compliance. Not only how they align with, but are a part of business goals.
Involve top management in decision-making related to quality:
Engage top management in the decision-making process. Seeking their input and making them feel invested in shaping the quality management system. It is important that the entire organization be ‘quality focused’ at all times. Not just when an audit or management review is approaching.
Consider the potential consequences of non-compliance with quality standards:
Emphasize the impact of not adhering to quality standards, such as regulatory penalties or reputational damage. This will underscore the necessity of the system’s implementation. This can be validated externally if need be. Auditors or consultants can assess quality processes and provide independent validation of a systems strengths of weaknesses.
It’s a common misconception that FDA De Novo content is very different from FDA 510k submission content, but is that true?
What do you think the De Novo content differences are?
Most people think the difference between a 510k and a De Novo is time and money. That conclusion is based upon a very important assumption: a 510k will not require clinical data, and a De Novo will require clinical data. That assumption is not always correct. 10-15% of 510k submissions include clinical data to support the performance claims, and last year our team submitted three De Novo submissions that did not include any clinical data. So what are the differences between a 510k and a De Novo content?
We use the same FDA eSTAR template for both types of FDA submissions, and on the first page of the eSTAR template, we identify if the submission is a 510k or De Novo. If we select De Novo, the eSTAR will be pre-populated with six unique De Novo content requirements covering four (4) different areas that are not found in a 510k. The six unique content requirements are:
Recommending a classification, providing a justification for that classification, and explaining what efforts were taken to identify a suitable 510k product code
Description of existing alternative practices or procedures used in diagnosing, treating, preventing, curing, or mitigating the disease or condition for which the IVD or device is intended
Providing a written benefit/risk analysis starting with the clinical benefits of your device
Efforts to identify a potential predicate (including identifying alternative practices, procedures, or even drugs)
Recommendations for FDA special controls for your new product code based upon the risks to health and the mitigation measures for each risk
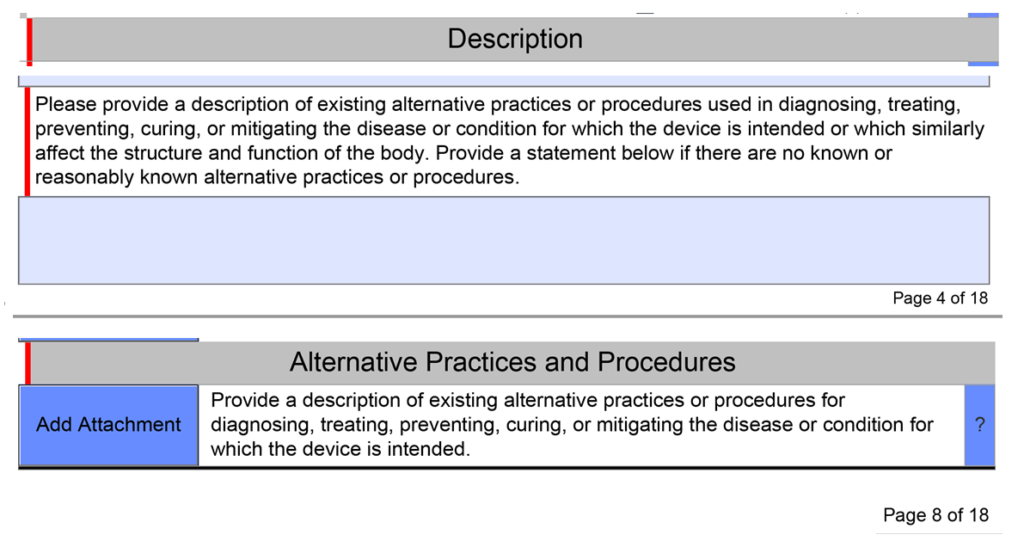
What alternative practices and procedures are currently available?
The unique De Novo content requirement is to provide a description of alternative practices and procedures for treatment or diagnosis of the same indications that you are proposing for your subject device. This is a subsection of the device description section in the FDA eSTAR template. Your should description should include other 510k-cleared products, drugs, and even products that have similar indications but are not identical. The description of alternative practices and procedures must also be attached as a document in the section for benefits, risks, and mitigation measures. To maintain consistency throughout your submission, you should create the document for attachment first and copy and paste the content into the text box at the end of the device description section.
You need to recommend a classification in your De Novo
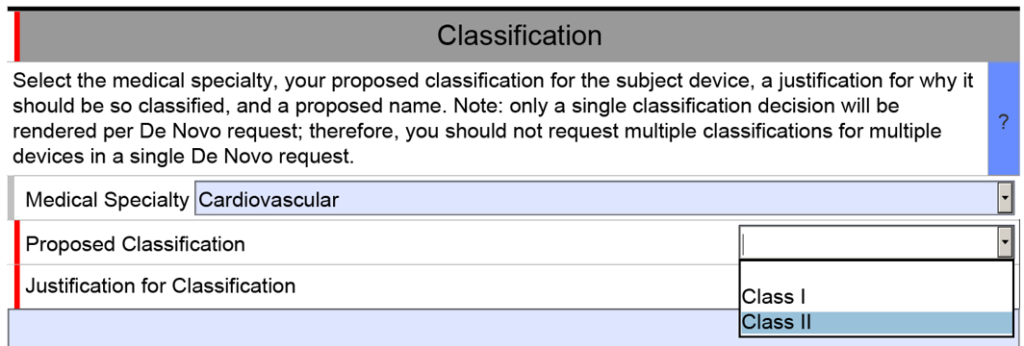
The unique De Novo content requirement is found in a section titled “Classification.” There is a shorter classification section included in 510k submissions, but the 510k version only has four cells. The first three are populated by selecting one of the options from a dropdown menu, and the fourth cell is only used if your subject device includes other product classification codes.
The De Novo version of the eSTAR is identical for the first row of the classification section, but then you must select a proposed product classification (i.e., Class 1 or Class 2) in accordance with FDA Classification Procedures (i.e., 21 CFR 860). The third cell is a text box for you to enter your justification for the proposed classification. Next, the FDA requires you to enter a proposed classification name. Finally, at the end of the classification section, the FDA requires that you provide a classification summary or reference to a previous NSE 510k submission.
A benefit-risk analysis is required in the De Novo content
For new devices, the FDA uses a benefit-risk analysis to decide if a device should be authorized for marketing in the USA. This process includes humanitarian device exemptions, De Novo applications, and Premarket Approval submissions. The FDA has a guidance document that provides guidance for FDA reviewers and the industry. The most important aspect is, to begin with, the benefits of the device and to provide a quantitative comparison of benefits and risks. Many De Novo submissions have been rejected because the submitter did not provide objective evidence of clinical benefits for the subject device.
The FDA guidance documents are helpful for creating a benefit-risk analysis, but you can also find information in the ISO/TIR 24971:2020–the guidance for the application of ISO 14971:2019. Our company also includes a template for a benefit/risk analysis as part of our risk management procedure (i.e., SYS-010).
What are your recommended Special Controls?
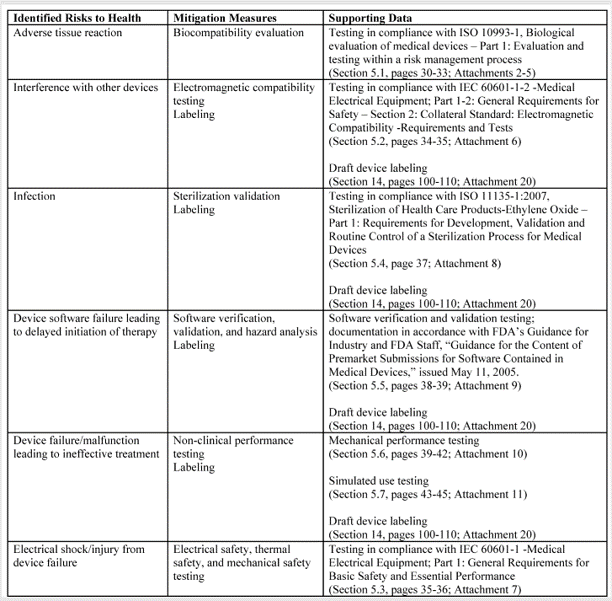
In FDA De Novo Classification Decision Summaries, there is a table provided that identifies the identified risks to health and the recommended mitigation measures for each risk category. In the FDA eSTAR, you are required to add a similar table for De Novo content. The only difference between the table in summary and the eSTAR is that the eSTAR table has a third column where the FDA wants you to reference the supporting data provided for each mitigation measure–including the document and page within the document. The FDA also provided an example table in the eSTAR, copied below.
The above table for the risks to health and mitigations needs to be translated into a list of recommended Special Controls for Class II devices. Since most De Novo applications are for Class II devices, you will need to convert each of your mitigations into a corresponding Special Control and type these controls into the text box provided in the FDA eSTAR.
What else is different from a 510k?
There are no additional mandatory elements that you need to include in a De Novo application, but there are several elements of a 510k submission that are not included in a De Novo. The most obvious of these sections is the Substantial Equivalence Comparison Table in the section labeled “Predicates and Substantial Equivalence.” Another difference is that you are more likely to need clinical data to support a De Novo application than for a 510k submission. It is also possible that subsequent 510k submissions for the same product code may not need to provide clinical data because the 510k process only requires a demonstration of substantial equivalence rather than clinical benefits outweighing risks to health. The FDA review time for a Traditional 510(k) varied between 190 and 210 days in 2022, while the De Novo review timeline averaged 390 days in 2022. Finally, the FDA user fees for 510k submissions are far less than those for a De Novo application.