Explanation of US FDA requirements for establishing a software development lifecycle that results in secure software.
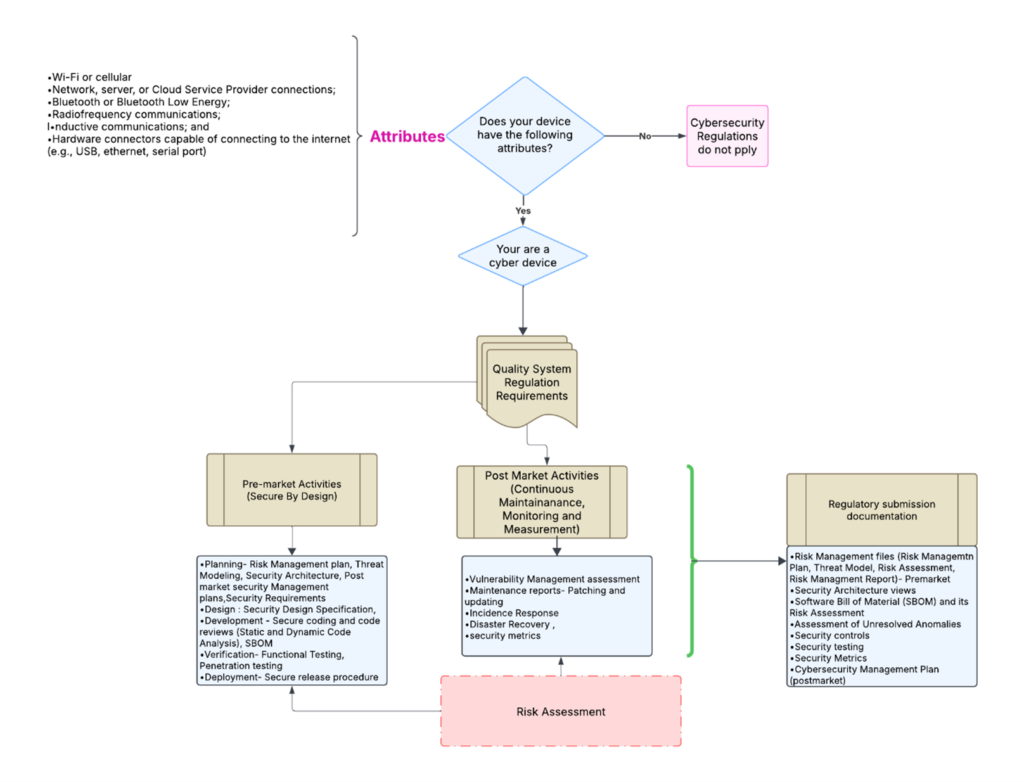
Iscybersecurityapplicabletoyourdevice?
As medical devices become more connected and threats evolve, the probability of a security breach increases. To address these growing concerns, the FDA has published 2023 guidance on “Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions” as an approach to strengthen the cybersecurity and safety of medical devices. The guidance requires manufacturers to integrate security as a fundamental aspect throughout the entire device lifecycle, from initial planning to decommissioning.
Your device is a cyber device if the following attributes apply
Wi-Fi or cellular
Network, server, or Cloud Service Provider connections.
Bluetooth or Bluetooth Low Energy.
Radiofrequency communications.
Inductive communications; and
Hardware connectors capable of connecting to the internet (e.g., USB, Ethernet, serial port)
How is secure software addressed by quality systems?
As the title of the guidance suggests, the FDA wants manufacturers to incorporate cybersecurity into their quality systems. These regulations will address both pre-market and post-market security requirements throughout the device’s life. This requirement ensures that a consistent design and labeling approach is employed throughout the industry as common practice.
Premarket Quality system regulation requirements must incorporate security-related phases, such as:
Planning
Design
Development
Testing
Deployment
Post-market Quality System requirements: To maintain robust cybersecurity, the manufacturers need to focus on the following key post-market areas for monitoring and maintenance:
Risk Management: Identify and mitigate risks by conducting regular vulnerability assessments.
Incident Response: Prepare an incident response plan for detecting, reporting, and responding to security breaches.
Software Updates: Regularly update and patch the software to ensure system security and integrity.
Reporting and Communication: Establish an effective communication strategy and policy for vulnerability disclosure.
Submission Requirements for Secure Software
The documentation requirements for regulatory submission will not be developed as a standalone.
To demonstrate that your device is secure and that it has been developed following a Secure Software Development Lifecycle (SSDLC), the manufacturers must include documents from both their pre-market and post-market security processes in their regulatory submissions to the FDA. The documents commonly include planning, secure coding, comprehensive risk assessment, and continuous security monitoring and management.
The documents for the regulatory submission include the following:
Threat Modeling
Security Architecture
Security risk management files
SBOM with EOL and LOS information
Safety and security risk Assessment of vulnerabilities in the OTS components identified in the SBOM.
Unresolved Anomalies for security impact
Security Metrics
Security controls
Security Testing
Security labeling
The flowchart below outlines a simplified process for determining pre-market and post-market requirements. It also shows what documents must be retrieved from the manufacturer’s quality systems for the regulatory submissions.
Data hazards occur when there is missing data, incorrect data, or a delay in data delivery to a database or user interface. This can occur with both software as a medical device (SaMD) and software in a medical device (SiMD). Missing data, incorrect data, and delays in data delivery cannot cause physical harm to a patient or user. Therefore, many medical device and IVD manufacturers state in their risk management file that their device or IVD has low risk or no risk. This is an incorrect software risk analysis because failure to meet software data or database requirements results in a hazardous situation, such as 1) an incorrect diagnosis or treatment, or 2) a delay in diagnosis or treatment. These hazardous situations can compromise the standard of care at best, or at worst, hazardous situations can result in physical harm–including death.
Where do you document these hazards?
Data hazards are documented in your software risk management file, but the data hazards are referenced in multiple documents. Usually data hazards will be referenced in a software hazard identification, the software risk analysis, software verification and validation test cases, and the software risk management report. Security risk assessments will also identify potential data hazards resulting from cybersecurity vulnerabilities that could be exploited.
How do you identify data hazards?
IEC 62304 is completely useless for the purpose of identifying data hazards. In Clause 5.2.2 of the standard for the software life-cycle process, the only examples of data definition and database requirements provided are form, fit, and function. IEC/TIR 80002-1, Guidance on the application of ISO 14971 to medical device software, is extremely useful. Specifically, in Table B1 the following potential data hazards are identified:
Mix-up of Data such as:
Associating data with the wrong patient
Associating the wrong device/instrument with a patient
Associating measurements with the wrong analyte
Loss of data resulting from events such as:
Connectivity failure or Quality of Service (QoS) issues
Incorrect data acquisition timing or sampling rates (i.e., measurement)
Capacity limitations during peak loads
Missing data fields (i.e., incorrect database configuration or database mapping)
Modification of data caused by:
Data entry errors during manual entry
Automated preventive maintenance
Reset of data
Rounding of data
Averaging of data
Patient data also presents a security risk. Access to data must be controlled for data entry, viewing, and editing. Other potential causes of data integrity issues include power loss, division by zero, overflow/underflow, floating point rounding, improper range/bounds checking, off-by-one, hardware failure, timing, and watch-dog time outs. In Table B2 the guidance provides additional examples of causes and risk control measures are recommended for each cause.
Data hazards associated with artificial intelligence (AI) and machine learning (ML)
There are also data hazards associated with AI/ML software. When an algorithm is developed there is a potential for improving the algorithm or making the algorithm worse. There is always a data bias resulting from the patient population selected for data collection and the clinical users that assign a ground truth for that data. Sometimes the data entered is subjective or qualitative data rather than objective, quantitative data. The sequence or timing of data collection can also impact the validity of data used for training an AI algorithm. AAMI CR 34971:2023 is a Guide on the Application of ISO 14971 to Machine Learning and Artificial Intelligence. That guidance identifies additional hazards associated with data and databases.
How are these hazards addressed in software requirements?
In IEC 62304, Clause 5.2.2, lists the content for twelve different software requirements (i.e., items A-L):
a) functional and capability requirements;
b) SOFTWARE SYSTEM inputs and outputs;
c) interfaces between the SOFTWARE SYSTEM and other SYSTEMS;
d) software-driven alarms, warnings, and operator messages;
e) SECURITY requirements;
f) user interface requirements implemented by software;
g) data definition and database requirements;
h) installation and acceptance requirements of the delivered MEDICAL DEVICE SOFTWARE at the operation and maintenance site or sites;
i) requirements related to methods of operation and maintenance;
j) requirements related to IT-network aspects;
k) user maintenance requirements; and
l) regulatory requirements.
These software requirements may overlap, because any specific cause of failure can result in multiple types of software hazards. For example, a loss of connectivity can result in mix-up of data, incomplete data, or modification of the data. Therefore, to ensure your software design is safe, you must carefully analyze software risks and evaluate as many test cases as you can to verify effectiveness of the software risk controls.
What is the best way to analyze data risks?
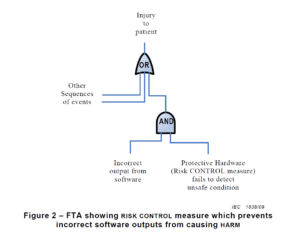
There are multiple risk analysis tools available to device and IVD manufacturers (e.g., preliminary hazard analysis, failure modes and effects analysis, and fault-tree analysis). Using a design failure modes and effects analysis (i.e., dFMEA) is the most common risk analysis tool, but a dFMEA is not the best tool for software risk analysis. There are two reasons for this. First, the dFMEA is a bottom-up approach that assumes you know all of the software functions that are needed–but you won’t. Second, the dFMEA will have multiple rows of effects for each failure mode because each cause of software failure can overlap with multiple software functions. Therefore, the best way to analyze data risks is a fault-tree analysis (i.e., FTA). The FTA is the best tool for analysis of software data hazards because you only need three fault trees: 1) Mix-up of data, 2) Loss of data, and 3) Modification of data. In each of these FTAs, all of the potential causes of software failure will be identified in the branches of the fault tree. Analyzing the fault tree structure, specifically the position of OR gates, can assist in software design. OR logic gates that can result in critical failures need additional software risk controls to prevent a single cause of software failure from creating a serious hazardous situation.
Copied from Section 6.2.1.5 from AAMI TIR 80002-1:2009
How to build a fault tree
The first step of your software risk analysis should be to identify data hazards. Once you identify the data hazards, you can build a fault tree for each of the three possible software failures: mix-up of data, 2) incomplete data, and 3) modification of data. Each data hazards can cause multiple software failure modes, but the type of logic gate will determine the outcome of that data hazard. An OR gate will result in software failures if there is just one hazardous event, while an AND gate requires at least two hazardous events to occur before the software failure will occur. The position of the OR/AND gate also impacts the potential for software failures.
Learn why you need to start with software validation documentation before you jump into software development.
When do you create software validation documentation for a medical device or IVD?
At least once a week, I speak with the founder of a new MedTech company that developed a new software application as a medical device (SaMD). The founder will ask me to explain the process for obtaining a 510(k), and they want help with software validation documentation. Many people I speak with have never even heard of IEC 62304.
Even though they already have a working application, usually, validation documentation has not even been started. Although you can create all of your software validation documentation after you create a working application, certain tasks are important to perform before you develop software code. Jumping into software development without the foundational documentation will not get your device to market faster. Instead, you will struggle to create documentation retroactively, and the process will be slower. In the end, the result will be a frustrating delay in the launch of your device.
What are the 11 software validation documents required by the FDA?
In 2005 the FDA released a guidance document outlining software validation documentation content required for a premarket submission. There were 11 documents identified in that guidance:
What the FDA guidance fails to explain is that some of these documents need to be created before software development begins, or your software validation documentation will be missing critical design elements. Therefore, it is important to create a software development plan that schedules activities that result in those documents at the right time. In contrast, four of the eleven documents can wait until your software development is complete.
Which of the software validation documents can wait until the end?
The level of concern only determines what documents the FDA wants to review in a submission rather than what documents are needed for a design history file. In fact, the level of concern (LOC) document is no longer required as a separate document in premarket submissions using the FDA eSTAR template because the template already incorporates the questions that document your LOC. The revision level history document is simply a summary of revisions made to the software during the development process, and that document can be created manually or automatically at the end of the process, or the revision level history can be a living document that is created as changes are made. The traceability matrix can also be a living document created as changes are made, but its only purpose is to act as a tool to provide traceability from hazards to software requirements, to design specifications, and finally to verification and validation reports. Other software tools, such as Application Lifecycle Management (ALM) Software, are designed to ensure the traceability of every hazard and requirement throughout the entire development process. Finally, unresolved anomalies should only be documented at the time of submission. The list may be incomplete until all verification and validation testing is completed, and the list should be the shortest at the time of submission.
What documentation will be created near the end of development?
The software design specification (SDS) is typically a living document until your development process is completed, and you may need to update the SDS after the initial software release to add new features, maintain interoperability with software accessories, or change security controls. The SDS can not begin, however, until you have software requirements and the basic architecture defined. The verification and validation activities are discrete documents created after each revision of the SDS and must therefore be one of the last documents created–especially when provided to the FDA as a summary of the verification and validation efforts.
Which validation documents do you need first?
At the beginning of software development, you need a procedure(s) that defines your software development process. That procedure should have a section that explains the software development environment–including how patches and upgrades will be controlled and released. If you don’t have a quality system procedure that defines your development process, then each developer may document their coding and validation activities differently. That does not mean that you can’t improve or change the procedure once development has begun, but we recommend limiting the implementation of a revised procedure when making major software changes and discussing how revisions will be implemented for any work that remains in progress or has already been completed.
When do the remaining software validation documents get created?
The remaining four software validation documents required for a premarket submission to the FDA are:
Software description
Software hazard analysis
Software requirements specification (SRS)
Architecture design chart
Your development process will be iterative, and therefore, you should be building and refining these four documents iteratively in parallel with your software code. At the beginning of your project, your design plan will need a brief software description. Your initial software description needs to include the indications for use, a list of the software’s functional elements, and the elements of your user specification (i.e., intended patient population, intended users, and user interface). If you are using lean startup methodology, the first version of your device description will be limited to a minimal viable product (MVP). The target performance of the MVP should be documented as an initial software requirements specification (SRS). This initial SRS might only consist of one requirement, but the SRS will expand quickly. Next, you need to perform an initial software hazard analysis to identify the possible hazards. It is important to remember that software hazards are typically hazardous situations and are not limited to direct physical harm. For each potential hazard you identify in your hazard analysis, you will need a software requirement to address each hazard, and each requirement needs to be added to your SRS. As your software becomes more complex by adding software features, your device description needs to be updated. As you add functions and requirements to your software application, your SRS will need updates too. Finally, your development team will need a tool to track data flow and calculations from one software function to the next. That tool is your architecture design chart, and you will want to organize your SRS to match the various software modules identified in your architecture diagram. This phase is iterative and non-linear, you will always have failures, and typically a team of developers will collaborate virtually. Maintaining a current version of the four software documents is critical to keeping your development team on track.
How do you perform a software hazard analysis?
One of the most important pre-requisite tasks for software developers is conducting a hazard analysis. You can develop an algorithm before you write any code, but if you start developing your application to execute an algorithm before you perform a software hazard analysis, you will be missing critical software requirements. Software hazard analysis is different from traditional device hazard analysis because software hazards are unique to software. A traditional device hazard analysis consists of three steps: 1) answering the 37 questions in Annex A of ISO/TR 24971:2020, 2) systematically identifying hazards by using Table C1 in Annex C of ISO 14971:2019, and 3) reviewing the risks associated with previous versions of the device and similar competitor devices. A software hazard analysis will have very few hazards identified from steps 1 and 2 above. Instead, the best resource for software hazard analysis is IEC/TR 80002-1:2009. You should still use the other two standards, especially if you are developing software in a medical device (SiMD) or firmware, but IEC/TR 80002-1 has a wealth of tables that can be used to populate your initial hazards analysis and to update your hazard analysis when you add new features.
How do you document your hazard analysis?
Another key difference between a traditional hazard analysis and a software hazard analysis is how you document the hazards. Most devices use a design FMEA (dFMEA) to document hazards. The dFMEA is a bottom-up method for documenting your risk analysis by starting with device failure modes. Another tool for documenting hazards is a fault tree diagram.
A fault tree is a top-down method for documenting your risk analysis, where you identify all of the potential causes that contribute to a specific failure mode. Fault tree diagrams lend themselves to complaint investigations because complaint investigations begin with the identification of the failure (i.e., complaint) at the top of the diagram. For software, the FDA will not allow you to use the probability of occurrence to estimate risks. Instead, software risk estimation should be limited to the severity of the potential harm. Therefore, a fault tree diagram is generally a better tool for documenting software risk analysis and organizing your list of hazards. You might even consider creating a separate fault tree diagram for each module of your software identified in the architecture diagram. This approach will also help you identify the potential impact of any software hazard by looking at the failure at the top of the fault tree. The higher the potential severity of the software failure, the more resources the software team needs to apply to developing software risk controls and verifying risk control effectiveness for the associated fault tree.
What is your company’s approach to qualifying a software service provider and managing software-as-a-service (SaaS) for cybersecurity?
The need for qualifying and managing your software service provider
Most of the productivity gains of the past decade are related to the integration of software tools into our business processes. In the past, software licenses were a small part of corporate budgets, and the most critical software tools helped to manage material requirements planning (MRP) functions and customer relationship management (CRM). Today, there are software applications to automate every business process. Failure of a single software service provider, also known as “Software-as-a-Service” or (Saas), can paralyze your entire business. In the past, business continuity plans focused on labor, power, inventory, records, and logistics. Today our business continuity plans also need to expand for the inclusion of software service providers, internet bandwidth, websites, email, and cybersecurity. This new paradigm is not specific to the medical device industry. The medical device industry has become more dependent upon its supply chain due to the ubiquity of outsourcing, and what happens to other industries will eventually filter its way into this little collective niche we share. With that in mind, how do we qualify and manage a software service provider?
Threats to software service providers (Kaseya Case Study)
Two years ago the WannaCry ransomware attack affected 200,000 computers, 150 countries, and more than 80 hospitals.
Kaseya isn’t a hospital. Kaseya is a software service provider company. So why is this example relevant to the medical device industry?
The ransomware attack on Kaseya was severe enough that both CISA and the FBI got involved, and it compromised some Managed Service Providers (MSPs) and downstream customers. This supply chain ransomware attack even has its own Wikipedia page. The attack prompted Kaseya to shut down servers temporarily. None of this is a critique of Kaseya or their actions. They were merely the latest high-profile victim of a cyberattack in the news. Now cybercriminals are attacking your supply chain. We want to emphasize the concepts and considerations of this type of attack as it pertains to your business.
What supplier controls do you require for a software service provider?
If you are a manufacturer selling a medical device under the jurisdiction of the U.S. FDA, you need to comply with 21 CFR 820.50 (i.e. purchasing controls). The FDA requires an established and maintained procedure to control how you are ensuring what your company buys meets the specified requirements of what you need. Many device manufacturers only consider suppliers that are making physical components, but a software service provider may be critical to your device if your device is software as a medical device (SaMD), includes software, or interacts with a software accessory. A software service provider may also be involved with quality system software, clinical data management, or your medical device files. Do you purchase software-as-a-service or rely upon an MSP for cloud storage?
You need to determine if your software service provider is involved in document review or approval, controlling quality records, Protected Health Information (PHI), or electronic signature requirements. You don’t need a supplier quality agreement for all of the off-the-shelf items your company purchases. For example, it would be silly to have Sharpie sign a supplier quality agreement because you occasionally purchase a package of highlighters. On the other hand, if you are relying upon Docusign to manage 100% of your signed quality records, you need to know when Docusign updates its software or has a security breach. You should also be validating Docusign as a software tool, and there should be a backup of your information.
21 CFR 820.50 requires that you document supplier evaluations to meet specified and quality requirements per your “established and maintained” procedure. The specified requirements for this supplier might include the following:
How much data storage do you need?
How many user accounts do you need?
Do you need unique electronic IDs for each user?
Do you need tech support for the software service?
Is the software accessed with an internet browser, is the software application-based, or both?
How much does this software service cost?
Is the license a one-time purchase? Or is it a subscription?
The quality requirements for a supplier like this may look more like these questions;
How is my information backed up?
Can I restore previous file revisions in the case of corruption?
How can I control access to my information?
Can I sign electronic documents? If yes, is it 21 CFR Part 11 compliant?
Does this supplier have downstream access to my information? (can the supplier’s suppliers see my stuff?)
Do I manage PHI? If so, can this system be made HIPAA compliant? What about HITECH?
What cybersecurity practices does this supplier utilize?
How are routine patches and updates communicated to me?
A risk-based approach to supplier quality management
ISO 13485:2016 requires that you apply a risk-based approach to all processes, including supplier quality management. A risk-based approach should be applied to suppliers providing both goods and services. For example, you may order shipping boxes and contract sterilization services. Both companies are suppliers, but in this example, the services provided by the contract sterilizer are associated with a much higher risk than the shipping box supplier. Therefore, it makes sense that you would need to exercise greater control over the sterilizer. Software service providers are much like contract sterilizers. SaaS is not tangible but the service provided may have a high level of risk and potential impact on your quality management system. Therefore, you need to determine the risk associated with SaaS before you can evaluate, control, and monitor a software service supplier.
First, you need to document the qualification of a new supplier. It would be nice if your cloud service provider had a valid ISO 13485:2016 certification. You would then have an objectively demonstratable record of their process controls and know that they are routinely audited to maintain that certification. They would also understand and expect to undergo 2nd party supplier audits because they operate in the medical device industry. Alternatively, a software service provider may have an ISO 9001:2015 certification. This is a general quality system certification that may be applied to all products or services. In the absence of quality system certification, you can audit a potential supplier. For some suppliers, this makes sense. However, many companies that are outside of the medical device industry do not even have a quality system because it is not required or typical of their industry. For the ones that do, though, you can likely leverage their existing certifications and accreditations.
Cybersecurity standards you should know
Most cloud service providers will not have ISO 13485 certification, because it is a quality management standard specific to the medical device industry. However, you might look for some combination of the following ISO standards that may be relevant to a software service provider:
ISO/IEC 27001 Information Technology – Security Techniques – Information Security Management Systems – Requirements
ISO/IEC 27002:2013 Information Technology. Security Techniques. Code Of Practice For Information Security Controls
ISO/IEC 27017:2015 Information Technology. Security Techniques. Code Of Practice For Information Security Controls Based On ISO/IEC 27002 For Cloud Services
ISO/IEC 27018:2019 Information Technology – Security Techniques – Code Of Practice For Protection Of Personally Identifiable Information (PII) In Public Clouds Acting As PII Processors
ISO 22301:2019 Security And Resilience – Business Continuity Management Systems – Requirements
ISO/IEC 27701:2019 Security Techniques. Extension to ISO/IEC 27001 and ISO/IEC 27002 For Privacy Information Management. Requirements And Guidelines
Does your software service provider have SOC reports?
The acronym “SOC” stands for Service Organization Control, and these reports were established by the American Institute of Certified Public Accountants. SOC reports are internal controls that an organization utilizes and each report is for a specific subject. SOC reports apply to varying degrees for SaaS and MSP Suppliers
The SOC 1 Report focuses on Internal Controls over Financial Reporting. Depending on what information you need to store on the cloud, this report could be more applicable to the continuity of your overall business than specifically to your quality management system.
The SOC 2 Report addresses what level of control an organization places on the five Trust Service Criteria: 1) Security, 2) Availability, 3) Processing Integrity, 4) Confidentiality, and 5) Privacy. As a medical device manufacturer, these areas would touch on control of documents, control of records, and process validation, among other areas of your quality system. Some suppliers may not share a SOC 2 report with you, because of the amount of confidential detail provided in the report.
The SOC 3 Report will contain much of the same information that the SOC 2 Report contains. They both address the five Trust Service Criteria. The difference is the intended audiences of the reports. The SOC 3 is a general use report expected to be shared with others or publicly available. Therefore, it doesn’t go into the same intimate level of detail as the SOC 2 report. Specifically, information regarding what controls a system utilizes is very brief if identified at all compared to the description and itemized list of controls in the SOC 2 Report.
Other ways to qualify and manage your software service provider
SOC reports will help paint a picture of the organization you are trying to qualify for. You will also need to evaluate the supplier on an ongoing basis. It is essential to know if the supplier is subject to routine audits and inspections to maintain applicable certifications and accreditations. For example, if their ISO certificate lasts for three years, you should know that you should follow up with your supplier for their new certificate at least every three years. On the other hand, if they lose certification, it may signify that the supplier can’t meet your needs any longer and you should find a new supplier.
There is a long list of standards, certifications, accreditations, attestations, and registries that you can use to help qualify a SaaS or MSP supplier. One such registry is maintained by Cloud Security Alliance (i.e. the CSA STAR registry). “STAR” is an acronym standing for Security, Trust, Assurance, and Risk. CSA describes the STAR registry in their own words:
“STAR encompasses the key principles of transparency, rigorous auditing, and harmonization of standards outlined in the Cloud Controls Matrix (CCM) and CAIQ. Publishing to the registry allows organizations to show current and potential customers their security and compliance posture, including the regulations, standards, and frameworks they adhere to. It ultimately reduces complexity and helps alleviate the need to fill out multiple customer questionnaires.”
Some of the questions your supplier qualification process should be asking about your SaaS and MSP suppliers include:
Why do I need this software service?
Which standards, regulations, or process controls need to be met?
What is required for qualifying suppliers providing SaaS or an MSP?
How will you monitor a software service provider?
ISO certification, SOC reports, and the CSA STAR registry are supplier evaluation tools you can use for supplier qualification and monitoring. When you use these tools, make sure that you ask open-ended questions instead of close-ended questions. Our webinar on supplier qualification provides several examples of how to convert your “antique” yes/no questions into value-added questions.
Your software service provider should be able to provide records and metrics demonstrating the effectiveness of their cybersecurity plans. Below are three examples of other types of records you might request:
Cloud Computing Compliance Controls Catalogue or “C5 Attestation Report”
System Security Plan for Controlled Unclassified Information in accordance with NIST publication SP 800-171
Privacy Shield Certification to EU-U.S. Privacy Shield or Swiss-U.S. Privacy Shield
The privacy shield certification may be especially important for companies with CE Marked devices in order to comply with the European Union’s General Data Protection Regulation (GDPR) orRegulation 2016/679.
A final consideration for supplier qualification is, “Who are the upstream suppliers?” It is essential to know if your new supplier or their suppliers will have access to Protected Health Information (PHI). Since you have less control of your supplier’s subcontractors, you may need to evaluate how your supplier manages their supply chain and which general cybersecurity practices your supplier’s subcontractors adhere to.
Additional cybersecurity, software validation, and supplier quality resources
For more resources on cybersecurity, software validation, and supplier quality management please check out the following resources:
This article defines software verification and validation for medical devices and provides an overview of CE Marking and 510k requirements. We also provide a link to our free download of a webinar on 510k software documentation.
Software Verification and validation is an essential tool for ensuring medical device software is safe. Software is not a piece of metal that can be put into a strain gauge to see if the code is strong enough not to break. That’s because software is intangible. You can’t see if it is in the process of failing until it fails. The FDA is concerned about software safety since many medical devices now include software. Software failure can result in serious injury or even death to a patient. This places significant liability on the device manufacturer to ensure their software is safe. One way to ensure software safety is to perform software verification and validation (V&V).
What is software verification and validation (V&V)?
Definitions of software verification and validation confuse most people. Which tasks are software verification? And which tasks are software validation? Sometimes the terms are used interchangeably. Even the FDA does not clearly define the meaning of these two terms for software. For example, in the FDA’s design control guidance documentthe following definitions are used:
“Verification means confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.”
“Validation means confirmation by examination and provision of objective evidence that the particular requirements for specific intended use can be consistently fulfilled.”
Specific intended use requirement…specified requirements…what is the difference? To understand the difference between the two terms, the key is understanding “Intended Use.” It is asking the question: “What is the software’s intended use?”
“Intended Use” is not just about a bunch of engineers sitting around a table coming up with really fresh ideas. “Intended Use” refers specifically to the patient/customer of the software and how it fulfills their needs (i.e., “User Needs”). Systematic identification of user needs is required, and the software must address the user needs. Identification of user needs is done through customer focus groups, rigorous usability studies, and consultation with subject matter experts such as doctors and clinicians providing expert insight.
“Intended Use” also ensures the safety of the process through the process of “Hazard Analysis,” whereby any hazard that could potentially cause harm to the patient/customer is identified. For each identified hazard, software requirements, software design, and other risk controls are used to make sure the hazard does not result in harm, or if it does, the severity of the harm is reduced as far as possible.
So if “Validation” ensures user needs are met, what is “Verification” and how does it apply to the software development process. “Verification” ensures that the software is built correctly based on the software requirements (i.e., design inputs), with regard to each task the software must perform (i.e., unit testing), during communication between software modules (i.e., integration testing) and within the overall system architecture (i.e., system-level testing). This is accomplished by rigorous and thorough software testing using prospectively approved software verification protocols.
CE Marking requirements for software verification and validation (V&V)
European CE Marking applications include the submission of a technical file that summarizes the technical documentation for the medical device. To be approved for CE Marking by a Notified Body, the device must meet the essential requirements defined in the applicable EU directive. The technical file must also include performance testing of the medical device in accordance with the “State of the Art.” For software, IEC/EN 62304:2006, medical device software – software life cycle processes, is considered “State of the Art” for the development and maintenance of software for medical devices. This standard applies to stand-alone software and embedded software alike. The standard also identifies specific areas of concern, such as software of unknown pedigree (SOUP). As with most medical device standards, the standard provides a risk-based approach for the evaluation of SOUP acceptability and defines testing requirements for SOUP.
FDA requirements for software verification and validation (V&V)
For 510k submissions to the US FDA, section 16 of the 510k submission describes the software verification and validation (V&V) activities that have been conducted to ensure the software is safe and effective. There are 11 documents that are typically included in this section of the submission for software with a moderate level of concern:
Level of Concern
Software Description
Device Hazard Analysis
Software Requirement Specification (SRS)
Architecture Design Chart
Software Design Specification (SDS)
Traceability Analysis
Software Development Environment Description
Verification and Validation Documentation
Revision Level History
Unresolved Anomalies (Bugs or Defects)
The FDA does not require compliance with IEC 62304 as the European Regulations do, but IEC 62304 is a recognized standard, and manufacturers must comply with all applicable parts of IEC 62304 if they claim to follow IEC 62304. The FDA also provides a guidance document for the general principles of software validation. The above requirements for software verification and validation documentation also apply to software as a medical device (SaMD).
Additional Resources
If you are interested in learning more about the documentation requirements for a 510k submission of a software medical device, please click here to download a free recording of our 510k software documentation webinar.
Medical Device Academy also has a new live webinar scheduled for Tuesday, January 5, 2016, @ Noon (EST). The topic is “Planning Your 2016 Annual Audit Schedule“. We are also offering this live webinar as a bundle with our auditor toolkit.
About the Author
Nancy Knettell is the newest member of the Medical Device Academy’s consulting team, and this is her first blog contribution to our website. Nancy is an IEC 62304 subject matter expert. To learn more about Nancy, please click here. If you have suggestions for future blogs or webinars on the topic of medical device software, please submit your requests to our updated suggestion portal.