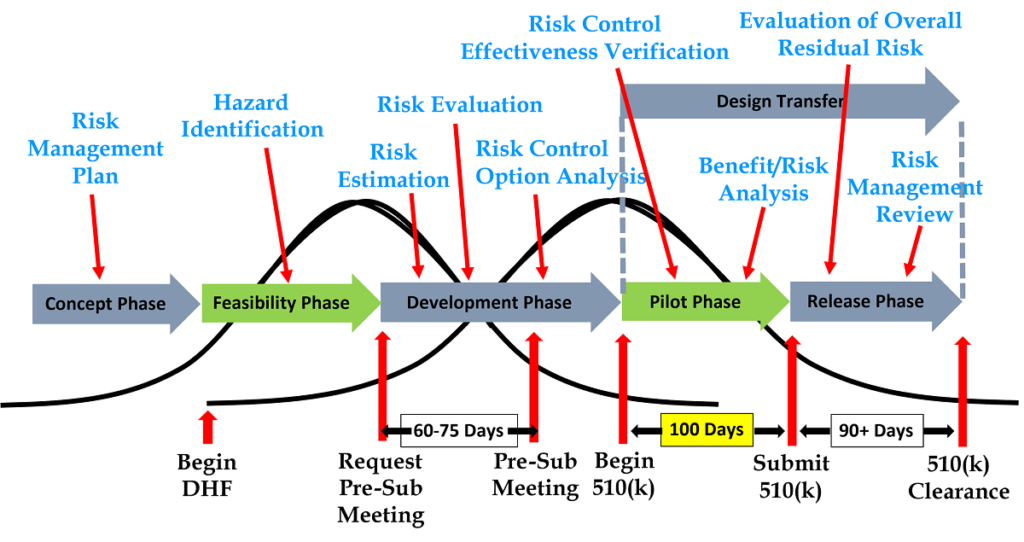
What’s new in the 2022 draft cybersecurity guidance?
On April 8, 2022, the FDA released a new draft cybersecurity guidance document to replace the 2018 draft that the industry does not support.
Why was the draft cybersecurity guidance created?
Due to the ubiquitous nature of software and networked devices in the medical industry, the impact of cybersecurity attacks is becoming more frequent and more severe. The WannaCry Ransomeware Attack is just one example of this global cybersecurity issue. The FDA is responding to the need for stronger cybersecurity controls by issuing a new draft cybersecurity guidance for 2022.

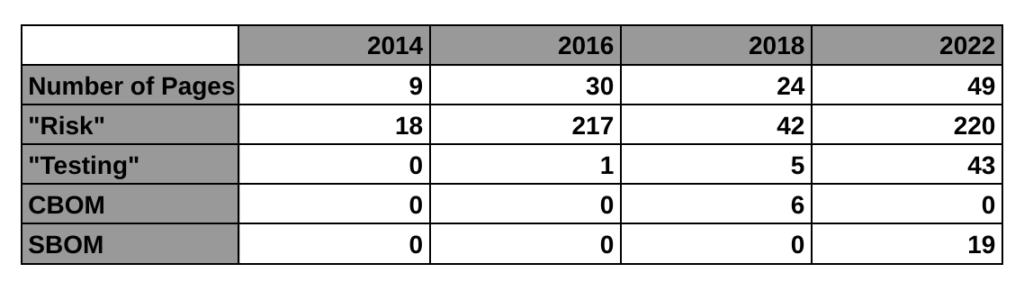
The first four paragraphs of the introduction explain why we need this, and WannaCry is mentioned in the second paragraph of the background section. This new guidance is only a draft, but this is the FDA’s third attempt at regulating the cybersecurity of medical devices. The first guidance was finalized in 2014. That’s the 9-page guidance we currently have in effect. The guidance mentions risk 11 times and there is no mention of testing requirements or a bill of materials (BOM). The 2018 draft guidance (24-pages) met with resistance from the industry for a lot of reasons. One of the reasons mentioned by Suzanne Schwartz in an interview is the inclusion of a cybersecurity bill of materials (CBOM). The industry felt it would be too burdensome to disclose all of the hardware elements that are related to cybersecurity. Therefore, the FDA rewrote the 2018 draft and released a new draft on April 8, 2022 (49-pages).

You might have expected the FDA to soften its requirements in the face of resistance from industry, but the new draft does not appear to be less robust. It is true that the CBOM was replaced by a software bill of materials (SBOM). However, the SBOM must be electronically readable and it must include:
- the asset(s) where the software resides;
- the software component name;
- the software component version;
- the software component manufacturer;
- the software level of support provided through monitoring and maintenance from the software component manufacturer;
- the software component’s end-of-support date; and
- any known vulnerabilities.
You can be sure that the medical device industry will view providing an SBOM as a hefty burden. After all, a machine-readable SBOM is more complex than UDI labeling requirements. An SBOM will not fit on the “Splash Screen” for anyone’s software application. Companies may provide documentation through the company website with a link in their software to that information. The format of the information could be in the “Manufacturer Disclosure Statement for Medical Device Security (MDS2).” However, MDS2 is a 349-line item Excel spreadsheet to be used as a checklist (i.e. quite a bit longer than the GUDID data elements spreadsheet), and it took the FDA eight years to complete the transition for the UDI Final Rule (i.e. 2013 – 2021).
The 2018 draft cybersecurity guidance document from the FDA required a cybersecurity bill of materials (CBOM). CBOM was defined as “a list that includes but is not limited to commercial, open source, and off-the-shelf software and hardware components that are or could become susceptible to vulnerabilities.” Therefore, the FDA’s change from a CBOM to an SBOM eliminated the requirement to disclose the hardware components. Despite the change in disclosure requirements, manufacturers will still be expected to monitor potential hardware vulnerabilities to cybersecurity attacks. It should also be noted that the language in the PATCH Act (a new bill submitted to the House of Representatives and to the Senate for ensuring the cybersecurity of medical devices) specifically requires manufacturers “to furnish a software bill of materials as required under section 524B (relating to ensuring the cybersecurity).”
Structure of the draft cybersecurity guidance
The 2022 draft cybersecurity guidance organizes the requirements into four major principles:
- cybersecurity as part of device safety and the quality system regulations
- designing for security
- transparency
- submission documentation
The draft cybersecurity guidance recommends the implementation of a Secure Product Development Framework (SPDF). However, there is not much detail provided in the guidance for a SPDF. In the past, the term for this type of process was referred to as a Secure Software Development Lifecycle (i.e. Secure SDLC). However, in February 2022, the NIST Computer Security Resource Center (CSRC) released version 1.1 of the Secure SDLC guidance which is now titled “Secure Software Development Framework (SSDF) Version 1.1: Recommendations for Mitigating the Risk of Software Vulnerabilities.” This guidance provides guidance on the implementation of best practices for reducing the risk of software vulnerabilities because existing standards for managing the software development lifecycle do not explicitly address software security (e.g. IEC 62304-1:2015). The SSDF recommends implementing a core set of high-level secure software development practices that can be integrated into your SDLC process. Your software development team will also require cybersecurity training.
Design for security is the second principle of the draft cybersecurity guidance
Under this new draft cybersecurity guidance, the FDA will be evaluating the cybersecurity of devices based on the ability of the device to provide and implement the following security objectives:
- Authenticity, which includes integrity;
- Authorization;
- Availability;
- Confidentiality; and
- Secure and timely updatability and patchability.
Transparency of cybersecurity information is for users
The draft cybersecurity guidance seeks to give device users more information pertaining to the device’s cybersecurity controls, potential risks, and other relevant information. This information will be in the form of an SBOM that is electronically readable. This information shall include disclosure of 1) known vulnerabilities or risks, 2) information to securely configure and update devices, and 3) communication interfaces and third-party software.
In addition to providing an SBOM, the FDA draft cybersecurity guidance includes requirements for cybersecurity labeling in section VI(A). There are 15 specific labeling requirements identified by the FDA for sharing with device users to improve the transparency of cybersecurity information. The first of these requirements is recommendations from the manufacturer for cybersecurity controls appropriate for the intended use environment (e.g., antimalware software, use of a firewall, password requirements). This first labeling requirement is identical to the 2018 draft guidance. Several of the other requirements are copied from the 2018 draft guidance, but others are new and/or reworded cybersecurity labeling requirements.
FDA Submission Documentation Requirements
The 2022 FDA draft cybersecurity guidance includes requirements for FDA submission documentation. Submission documentation must include a security risk management plan and report. The draft cybersecurity guidance explains on page 13 (numbered 9) that “performing security risk management is a distinct process from performing safety risk management as described in ISO 14971:2019.” Therefore, instead of using your safety risk management process, your software development team will need to have a different risk management process for software security. Details on the content for security risk management plans and reports can be found in AAMI TIR57:2016 – Principles for medical device security—Risk management. Appendix 2 also provides guidance for the inclusion of a) call flow diagrams, and b) information details for an architecture view.
Cybersecurity testing requirements for your FDA submission
The biggest impact of this new draft guidance may be the requirement for testing. The 2014 guidance has no testing requirement, the 2018 draft guidance mentioned testing 5 times in a few bullet points, but this new draft guidance mentions testing 43 times. The testing requirements for cybersecurity risk management verification include:
- Security requirements
- Threat mitigation
- Vulnerability testing
- Penetration testing
This guidance also includes a paragraph with multiple bullets of requirements for each of the four types of testing. This would essentially double the size and scope of the current software section for a 510k submission, and manufacturers will need to create new procedures and templates for their cybersecurity risk management process. For example, penetration testing requirements include the following elements:
- Independence and technical expertise of testers,
- Scope of testing,
- Duration of testing,
- Testing methods employed, and
- Test results, findings, and observations.
Differences between the cybersecurity guidance documents
The following table provides a high-level overview comparing the four cybersecurity guidance documents released by the FDA, including the 2016 guidance on post-market management of cybersecurity:
Vulnerability management plans
The FDA draft cybersecurity guidance document also has a requirement for manufacturers to develop a plan for identifying and communicating vulnerabilities to device users after the release of the device. The FDA requires this plan to be included in your device submission. The vulnerability management plan should include the following information (in addition to the requirements of the 2016 guidance for postmarket cybersecurity management):
- Personnel responsible;
- Sources, methods, and frequency for monitoring for and identifying vulnerabilities (e.g. researchers, NIST NVD, third-party software manufacturers, etc.);
- Periodic security testing to test identified vulnerability impact;
- Timeline to develop and release patches;
- Update processes;
- Patching capability (i.e. rate at which update can be delivered to devices);
- Description of their coordinated vulnerability disclosure process; and
- Description of how manufacturer intends to communicate forthcoming remediations, patches, and updates to customers.
What’s the next step for the draft cybersecurity guidance?
In March the “Protecting and Transforming Cyber Health Care Act of 2022 (PATCH Act)” was introduced to the House of Representatives and the Senate. The goal of the PATCH Act is to enhance medical device security by requiring manufacturers to create a cybersecurity risk management plan for monitoring and addressing potential postmarket cybersecurity vulnerabilities. The FDA seeks comments on the draft cybersecurity guidance through July 7, 2022. Given the support of the new bill in the House of Representatives and Congress, it is likely that the FDA will get the support it needs for this new guidance.
What’s new in the 2022 draft cybersecurity guidance? Read More »