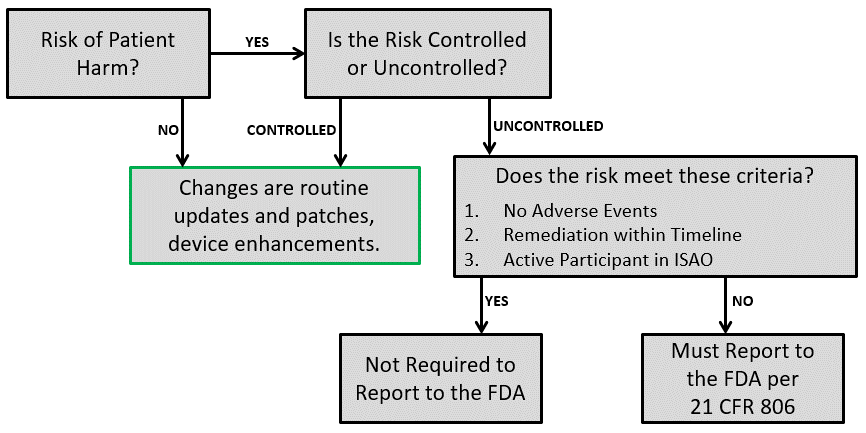
Purchasing Controls and Supplier Qualification
This article identifies the requirements for purchasing controls and supplier qualification procedures, as well as best practices for implementation.

Purchasing Controls
Sourcing suppliers in the medical device industry is not as simple as going on the internet and finding your material and purchasing it. As part of a compliant quality management system, purchasing controls must be in place to ensure that quality products and materials are going into your device and that any service providers that your company uses in the production of your product or within your quality management system are qualified.

ISO 13485 Requirements
In light of that, ISO 13485:2016 sections 7.4.1 Purchasing process, 7.4.2 Purchasing information, and section 7.4.3 Verification of purchased product outline the purchasing controls for medical device manufacturers. The following are requirements for the evaluation and selection of suppliers:
- The organization must have established criteria for the evaluation and selection of suppliers.
- The criteria need to evaluate the supplier’s ability to provide a product that meets the requirements.
- It needs to take into consideration the performance of the supplier.
- It must consider the criticality and the effect that the purchased product may have on the quality of the medical device.
- The level of supplier assessment and monitoring should be proportionate to the level of risk associated with the medical device.
Maintaining Purchasing Controls
To start, in the most basic sense, purchasing controls involve procedures that ensure you are only purchasing from suppliers who can meet your specifications and requirements. The best way to keep track of your qualified suppliers is to maintain an Approved Supplier List (ASL). You should only purchase products or services that affect your product or quality management system from companies on the ASL (you would not necessarily need to qualify things like office supplies or legal assistance through purchasing controls).
When used effectively, the Approved Supplier List can be a great tool to manage the key facets of purchasing control and keep track of supplier monitoring. Items that you can capture on the ASL include:
- Supplier Name
- Scope of Approved Supplies
- Contact Information
- Status of Approval (Approved, Pending, Unapproved, etc.)
- Qualification Criteria
- Supplier Certification and expiry dates
- Monitoring Requirements/Activities
- Date of Last Review
- Date of Next Review
The first step in your purchasing procedure should involve checking to see if the supplier is under active approved status on the ASL. The second step will be to ensure that you are purchasing an item/service that is within the scope of approval of that supplier. If you have not approved the supplier, or the intended purchase is beyond the scope of that supplier, your purchaser will need to go through the necessary channels to add the supplier to the ASL or modify their scope on the ASL.
Supplier Qualification Criteria
As required by the FDA, the level of supplier assessment should be proportionate to the level of risk associated with the medical device. The FDA is not prescriptive about the use of specific qualifications or assessments for different types of suppliers, so that is up to your company to determine. This is a somewhat grey area but based on years working with companies and suppliers, as well as participating in FDA and ISO 13485 audits, there are some general expectations of vendor qualifications that we have observed and would recommend.
It is good practice to have a form or template that guides your supplier evaluation process. Using input from engineering and QA to first determine the level of risk and the requirements of that supplier, and then base your qualification plan on that information. If you have a higher risk supplier who may be supplying a critical component to your device, or providing a critical service such as sterilization, then your qualification process will be much more involved.
Here is an example of two different levels of criteria based on the type of supplier (the intent is not for the following items to be rules, and your company is responsible for determining the adequate acceptance criteria for suppliers, but this is a general example of what you may expect).
- Critical Custom Component Supplier
- ISO 13485 Certification
- On-site audit of supplier’s facility
- References
- Provides Certificates of Analysis (CoA)
- A written agreement that the supplier will communicate with the company regarding any changes that could affect their ability to meet requirements and specifications.
- You validate a production sample, and it meets requirements
- Non-Critical Consumable Supplier
- Product available that meets the needs of the company.
- An associate has previously used by an associate who recommends the supplier.
- Adequate customer service returns allowed.
Additional Function of Supplier Evaluation Forms
The supplier evaluation form can also be used as the plan to assign responsibility and track completion and results during the initial evaluation. It can also include the plan for ongoing monitoring and control of the supplier. This evaluation form should be maintained as a quality record, and auditors will frequently ask to see supplier evaluations.
Are Supplier Audits Required as Purchasing Controls?
Also valuable, supplier audits may be included as part of an evaluation plan for a new supplier, the change of scope of a supplier, a routine audit as part of ongoing monitoring, or as part of a nonconformity investigation of a high-risk product. While it is not required by ISO 13485, nor does the FDA does specify in the CFR that you must audit suppliers, it is a very good idea to audit your critical suppliers. If an auditor or FDA inspector sees evidence that your current purchasing controls are inadequate, performing supplier audits may be forced as a corrective action.
Beyond that, you can gain so much value, and gather countless clues and important information in an audit that you just cannot get without visiting your critical supplier. You can see where they plan to/are making/cleaning/sterilizing/storing your product. Talk to the people on the line, are they competent and trained? Does the company maintain their facility well? How secure is it? Do they maintain adequate records and traceability? Have there been any nonconformities relating to your product that have been detected? Etc.
Supplier audits should also include evaluation of the procedures, activities, and records of the supplier that could have an impact on the product or service they are providing your company. If it is not the first audit of the company, you should be sure to review the previous audit report findings and ensure the company has addressed any nonconformities, review supplier performance data, information about any changes that may have occurred at the supplier since your last visit, etc.
Record Maintenance and Ongoing Evaluation of Suppliers
No matter the method of supplier qualification, it is best practice to maintain supplier files that contain useful information relative to the supplier that may include:
- The original supplier qualification form
- Supplier certificates
- References
- Audit reports
- Subsequent performance evaluations
- Expanded scope qualifications
- Supplier communications
- Current contact information
- Copies of any non-conforming material reports related to the supplier, etc.
ISO 13485 requires monitoring and re-evaluation of suppliers, and maintaining detailed supplier files will assist in meeting this requirement, and will help in the feedback system to identify and recurring problems or issues with a supplier. On a planned basis, whether that is annually, or every order (dependent on the criticality of the product), your company should conduct a formal supplier evaluation to determine whether the supplier has continued to meet requirements – In general, annual supplier reviews are standard. Additionally, you must specify this frequency in your procedure (auditors will look for what period you specify in your procedure, and then will check your ASL to make sure all of your suppliers have been reviewed within that timeframe).
During the supplier evaluation, if you find there have been issues, you need to determine and weigh the risks associated with staying with that supplier, and document that in the supplier file. If you determine the supplier should no longer be qualified, then you must also indicate on the ASL that the company no longer approves of the supplier.
Making the Purchase
When you have verified your supplier is approved on the ASL, you are authorized to purchase a product. Engineering is usually responsible for identifying the product specifications, requirements for product acceptance, and adequacy of specified purchasing requirements before communication to the supplier. The specifications may be in the form of drawings or written specifications. Additional information communicated to the supplier should also include, as applicable, an agreement between your company and the supplier that the supplier will notify you before the implementation of changes relating to the product that could affect its ability to meet specified purchasing requirements. When the first batch of product is received from a particular supplier, it is a good idea to verify that the product performs as intended before entering into production with new material or components.
Supplier Nonconformity
From time to time, you may encounter issues with a supplier. Sources of nonconformity include incoming inspections, production nonconformities, final inspection, or customer complaints. You must notify your supplier of the nonconformity and record their response and assessment. Depending on the level of criticality of the vendor, it is reasonable to require them to perform a root cause analysis to determine and alleviate the cause of failure. You should also request documentation of an effectiveness check to ensure the supplier has taken corrective actions. You should maintain copies of supplier nonconformity reports in the supplier file, and discuss nonconformities during ongoing supplier evaluations.
If the supplier does not cooperate or fails to address the nonconformity in an acceptable manner, or if there is a pattern of nonconformities with the vendor, then you should disqualify the supplier, and indicate that the supplier is “not approved” on the ASL.
Purchasing Controls Procedures You Might Need
Medical Device Academy developed a Supplier Qualification Procedure, Purchasing Procedure, and associated forms that will meet purchasing controls regulatory requirements for ISO 13485:2016 and 21 CFR 820.50. These procedures will help you ensure that goods and services purchased by your company meet your requirements and specifications. If you have any questions or would like help in developing a custom procedure or work instructions that meet your company’s unique needs, please feel free to email me or schedule a call to discuss.
Purchasing Controls and Supplier Qualification Read More »