Risk Management Requirements – 510k vs DHF
What are the differences between 510k risk management requirements and risk management requirements for your Design History File (DHF)?
Risk management requirements integration with design
Last week I presented a free webinar on how to combine risk management with design controls when planning to submit a 510k. Many questions were asking what the design control and risk management requirements are for a 510k.

What are the 510k design control requirements?
There is no specific part of the regulations stating what the 510k design control requirements are. However, some aspects of the DHF are required as 510k design control documentation, but not necessarily in the exact form as maintained in the DHF. For example, Design Inputs and Design Outputs are presented as applicable recognized standards and design specifications, while others will remain precisely the same (i.e., verification and validation test reports).
What are the Risk Management Requirements in a 510k?
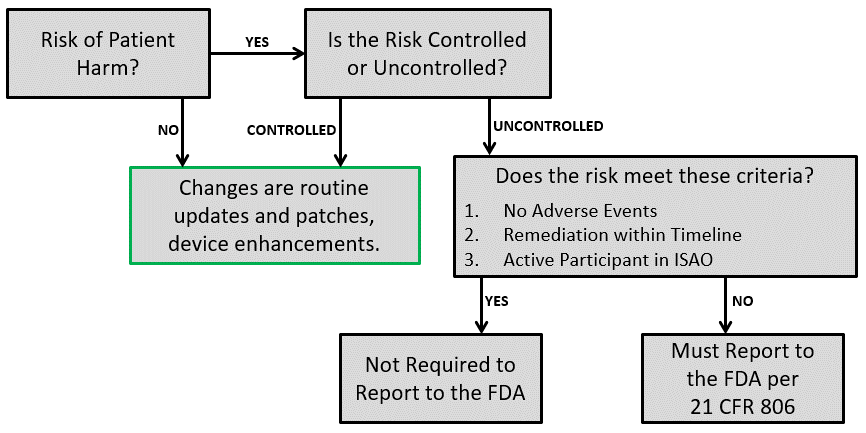
For 510k submissions, the only risk management requirements are the inclusion of risk documentation for devices containing software of at least moderate level risk. There are some exceptions to this as well, though, based on a few special control guidance documents—especially when the submission type is an abbreviated 510k. This is article identifies which of the DHF and RMF elements are 510k design control requirements and 510k risk management requirements.
Quality system requirements for design controls
Design Controls are identified in 21 CFR 820.30. Every manufacturer of any Class II or Class III devices and certain Class I devices (Class I devices with software, tracheobronchial suction catheters, surgeon gloves, protective restraints, radionuclide applicators, radionuclide teletherapy devices) need to control design per this regulation. The requirement for a Design History File is item j) and states:
“Each manufacturer shall establish and maintain a DHF for each type of device. The DHF shall contain or reference the records necessary to demonstrate that the design was developed following the approved design plan and the requirements of this part.”
The “requirements of this part” refer to the other bullets in 21 CFR 820.30 which can be summarized as:
a) Establish and maintain procedures to control the design of a device.
b) Design and Development Planning – Each manufacturer shall establish a plan that describes the design and development activities and defines responsibilities for implementation.
c) Design Inputs – Manufacturers need to ensure design requirements relating to a device are appropriate and address the intended use of the device.
d) Design Outputs – Design outputs need to be documented in terms that allow an adequate evaluation of conformance to design input requirements. Design outputs that are essential for the proper functioning of the device should be identified.
e) Design Review – Formal documented reviews of design results should be planned and conducted at appropriate stages of device development.
f) Design Verification – Design verification confirms that the design output meets the design input requirements.
g) Design Validation – Design validation shall be performed under defined operating conditions on initial production units or their equivalents. It shall ensure that devices conform to defined user needs and meet the intended use of the device.
h) Design Transfer – Design transfer documentation shall ensure that the device design is correctly translated into production specifications.
i) Design Changes – changes should be identified, documented, validated/verified, reviewed, and approved before their implementation.
The Design History File is intended to be a repository of the records required to demonstrate compliance with your design plan and design control procedures. While companies are required to create and maintain this documentation according to the FDA regulation, not all of the documentation will be reviewed as part of the 510k. The following table compares the elements that comprise a DHF with the 510k design control requirements.
| DHF Element | 510k Design Control Requirements |
| Design Plan | Not Required |
| User Needs & Design Inputs |
Declaration of Conformity User needs are design requirements that require design validation (e.g., adequacy of user training, and safety/performance of the device for the indications for use). Some design inputs will appear in the form of standards in the FDA eSTAR template. If you are declaring conformity with these standards, a Declaration of Conformity is automatically created in the FDA eSTAR template. |
| Design Outputs |
Device Description (Section 11) The Device Description lists the specifications of the device, and your Design Outputs document will help populate the Device Description. This can include drawings, pictures, or written specifications that describe your device. |
| Labeling |
Proposed Labeling (Section 13) The labeling is usually considered part of the Design Outputs within the DHF and is included specifically in the labeling section of the 510(k) submission. This includes both the Instructions for Use and any Package Labeling. |
| Verification and Validation Protocols |
Not Required You do not have to include the protocols, but the reviewer may ask to see them if they have any questions when reviewing the reports. |
| Verification and Validation Reports |
Sterilization (Section 14) Biocompatibility (Section 15) Software (Section 16) Electrical Safety and EMC (Section 17) Bench Performance Testing (Section 18) Animal Performance Testing (Section 19) Clinical Performance Testing (Section 20) Of course, not all of these sections will be applicable to every device. Still, you should include all relevant validation test reports within your submission in the appropriate part of the 510k. Typically, each of these sections will have a cover sheet that outlines the reports that are included within the section, and then you can just include the report from the DHF in its entirety behind the cover sheet in that section. |
| Process Validation | Only required for sterilization validation typically, but there are exceptions for novel materials and coatings |
| Work Instructions | Not Required for 510k |
| Design Review Meeting Minutes | Not Required for 510k |
| Design Trace Matrix | Only required for software |
| Risk Management File | Sometimes – See Risk Management File Table Below |
| Post-Market Surveillance Plan | Not Required, but a few exceptions for high-risk devices |
| Clinical Data Summary | Required only if used to demonstrate safety and efficacy |
| Regulatory Approval | It Will result from 510k Clearance, so nothing is to be included in the 510k submission. |
510k Risk Management Requirements
Regarding the FDA regulations for risk management, there is a requirement under the Design Validation section of 21 CFR 820.30 that states:
“Design validation shall include software validation and risk analysis, where appropriate.”
For FDA compliance and CE Marking, both recognize ISO 14971 as the standard for risk management. FDA recognizes ISO 14971:2007 whereas EN ISO 14971:2012 is the European National version for CE Marking. Rob Packard wrote an article describing the contents of the risk management file as well as the specific differences in the requirements between the FDA and CE Marking with regard to ISO 14971.
For your 510k submission, the FDA only requires risk management documentation to be included if the product contains software, and the risk is at least a level of “moderate concern”. There are some other cases when risk management is required by special controls guidance documents, but even when it is required, you only have to submit your risk analysis. The table below describes the risk management requirements in greater detail.
| RMF Element | 510k Risk Management Requirement |
| Risk Management Plan | Not Required |
| Hazard Identification |
510ks with Software Only (Section 16) Hazard Identification is only required for devices that have a software component. It is not required for most other devices. |
| Risk Assessment |
510(k)s with Software (Section 16) Certain Special Controls Guidance The Risk Assessment is only required to be included in your device contains software, or if a special controls guidance document specifically requires a risk assessment. It is not required for other 510ks. |
| Risk Control Option Analysis | Software and Certain Special Controls Guidance |
| Risk Control Verification and Validation |
Sterilization (Section 14) Biocompatibility (Section 15) Software (Section 16) Electrical Safety and EMC (Section 17) Bench Performance Testing (Section 18) Animal Performance Testing (Section 19) Clinical Performance Testing (Section 20) This will not be any additional or special documentation specific to Risk Management and was already included in the DHF breakdown above. Still, the verification and validation also relate to risk management in ensuring that the risks have been adequately mitigated. |
| Risk-Benefit Analysis |
Not Required for 510(k) Risk-Benefit analyses are only required for De Novo applications, Humanitarian Device Exemptions, and PMAs. |
| Informing Users and Patients of the Risks |
Labeling (Section 13) Part of the risk management will appear in the Labeling section of the 510k as warnings, contraindications, and precautions within the Instructions for Use and Package Labeling. |
| Risk Management Report | Not Required |
Special Controls Guidance Documents with Risk Management Requirements
Your first step in preparing your 510k submission is to search the FDA Guidance Document Database to determine if there is an applicable guidance document for your device. You can read another blog we wrote to explain Special Controls Guidance documents, and how to determine if one applies to your device. The following list provides examples of Class II Special Controls Guidance documents that require risk analysis to be included within the 510k:
- Class II Special Controls Guidance Document: Instrumentation for Clinical Multiplex Test Systems – Guidance for Industry and FDA Staff
- Class II Special Controls Guidance Document: Transcutaneous Air Conduction Hearing Aid System (TACHAS); Guidance for Industry and FDA
- Class II Special Controls Guidance Document: Pharmacy Compounding Systems; Final Guidance for Industry and FDA
- Guidance for Industry and FDA Staff – Class II Special Controls Guidance Document: Full Field Digital Mammography System
When there are 510k risk management requirements, the special controls guidance document will typically state, “We recommend that the summary report contain:
An identification of the Risk Analysis method(s) used to assess the risk profile in general as well as the specific device’s design and the results of this analysis. (Refer to Section 6 for the risks to health generally associated with the use of this device that the FDA has identified.)
Discussion of the device characteristics that address the risks identified in this class II special controls guidance document, as well as any additional risks identified in your risk analysis.”
The special controls guidance will also identify risks to health that have been identified for products of that type, which you should be sure to include in your risk analysis as appropriate.
More Information on Design Control and Risk Management Requirements
Hopefully, you are now able to determine which elements of your DHF are 510k design control requirements and which elements of your RMF are 510k risk management requirements. If you would like more information about how to implement design controls and risk management within your product development process, please consider registering for one of our training webinars:
- Risk Management Training Webinar
- Design Control Training Webinar
- Combining Product Risk Management with Design Controls
If you need any further information or specific assistance with your 510k submission, please feel free to schedule a call with Rob Packard to answer any of your medical device regulatory questions.

Risk Management Requirements – 510k vs DHF Read More »