7 Steps to writing an FDA 483 response
Responding in 15 business days is one of 7 steps on how to write an FDA 483 response, but do you know what should be in your response? When an FDA investigator has an inspection observation, the investigator issues an FDA 483. “Form 483” is the FDA form number. If your company receives an FDA 483, it is critical to understand how to write your FDA 483 response in order to avoid a Warning Letter. In the words of a former FDA investigator, “Many, many times I have seen an [Official Action Indicated (OAI)] classified inspection that had been recommended for a Warning Letter by the compliance branch be set aside based upon the response of the firm.”
When an FDA investigator has an inspection observation, the investigator issues an FDA 483. “Form 483” is the FDA form number. If your company receives an FDA 483, it is critical to understand how to write your FDA 483 response in order to avoid a Warning Letter. In the words of a former FDA investigator, “Many, many times I have seen an [Official Action Indicated (OAI)] classified inspection that had been recommended for a Warning Letter by the compliance branch be set aside based upon the response of the firm.”

The best way for your company to write a FDA 483 response is to provide a brief cover letter and to use your CAPA process. Every 483 inspection observation needs to be addressed in the FDA 483 response as a separate CAPA. Make sure that your response includes the following seven steps below:
- respond within 15 business days (earlier is better)
- use your CAPA form and a cover letter–instead of a memo
- document the investigation that was conducted with a concisely stated root cause
- identify containment measures and corrections to address each specific observation by the FDA inspector
- identify corrective actions planned and the date(s) you expect to complete implementation
- Include documentation of containment, corrections and corrective actions that are completed at the time you submit the response
- follow-up with a memo confirming that all the corrective actions are complete and include all related documentation–including training for any new procedures or any new corrective actions that warranted training
Your FDA 483 response is required in less than 15 business days
The FDA has always involuntarily required a medical device firm, or any firm under FDA jurisdiction that received an FDA 483, to provide a written FDA 483 response to the District Office within 15 business days. As of two years ago (http://www.gpo.gov/fdsys/pkg/FR-2009-08-11/pdf/E9-19107.pdf), it became mandatory that the Agency must receive a FDA 483 response within 15 business days, or an automatic Warning Letter is issued. You need to respond aggressively to FDA 483s with corrective actions, and submit your response early. The FDA has also modified the format of the response to require email responses.
Use your CAPA forms instead of a memo.
I have asked several former FDA investigators whether they would prefer to see firms submit responses in memo format, or by using their CAPA forms and a cover letter. Some told me that they prefer to see firms use their CAPA forms, while others don’t seem to have a preference. Nobody from the FDA has ever indicated a preference for a memo. I see no point in doubling your work and risking transcription errors. If you have an electronic system that does not have an easy-to-follow output format, go ahead and copy-and-paste the information from your electronic database to your memo. If the CAPA system output is easy to follow, just use a cover letter and copies of the forms.
Document the investigation and root cause
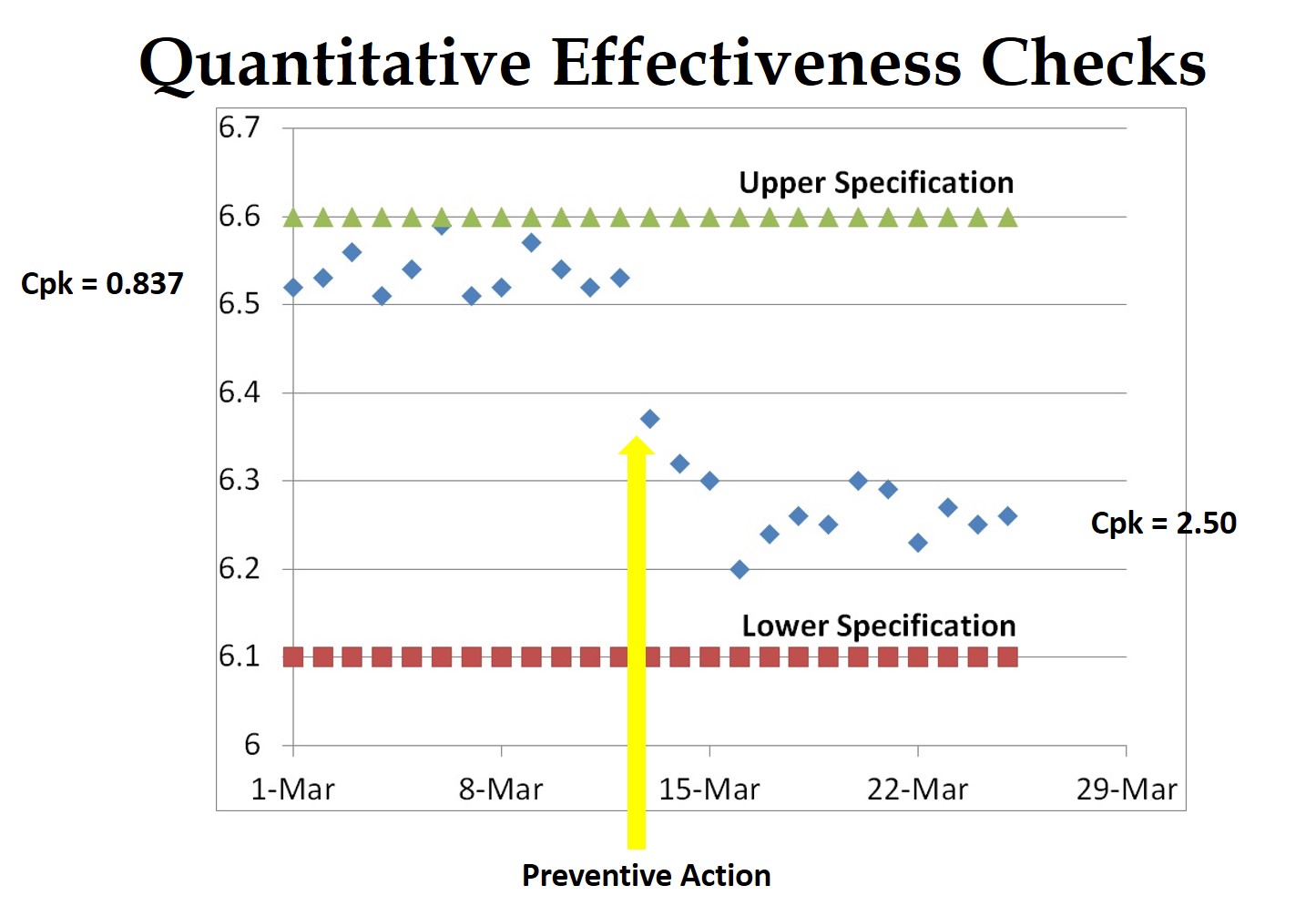
This is definitely my pet-peeve, but a one-sentence “root cause” is not enough for an FDA 483 response. Regardless of whether I am doing a mock-FDA inspection, an internal audit, or a supplier audit–I expect you to document how you determined the root cause (http://robertpackard.wpengine.com/five-tools-for-conducting-root-cause-analysis/). If it’s trivial and obvious, then it must have been something important, or I would not have written a nonconformity. Therefore, you should be looking beyond the immediate scope of the FDA 483 to ensure that a similar problem cannot occur elsewhere. In the language of the FDA, this is a preventive action, because you are preventing occurrence with another process or product. Most ISO certification auditors are purists, and they won’t accept this as a preventive action. You will have to show the purists something special–maybe from your data analysis.
Don’t forget containment and correction
For every 483 observation, including the subparts, you need to identify if immediate containment is necessary and how you can correct the problem. Whenever possible, you should attempt to implement the containment and corrections during your FDA inspection. It would be fantastic to give the FDA inspector a copy of the new CAPA you initiated during the audit. The new CAPA would identify containment and corrections that have been or will be implemented–including any nonconformity(s) you initiated to quarantine product. You may still get an FDA 483 inspection observation, but you are likely to convert a possible Official Action Indicated (OAI) into a Voluntary Action Indicated (VAI). You can also modify the CAPA wording later in your FDA 483 response to include a cross-reference to the FDA 483 and quote the exact wording the inspector uses.
Explain the corrective action plans and timelines
Clarity, brevity, and realistic plans are critical in this section of your response. I prefer a table that looks like the example shown below.

Show the FDA you have already taken action in your FDA 483 response
Whenever possible, you want to show the FDA that you are taking action without delay. If you revised the SOP for MDRs and scheduled a group training for July 15, then you should provide the FDA a copy of the revised procedure and a copy of the agenda for your planned training session. The only caution is to only commit to actions you are certain you will implement. You can always do more, but it will be much harder to explain why you did not implement an action you submitted in your FDA 483 response.
Follow-up with a second FDA 483 response before the FDA asks for it
The FDA’s compliance office will be looking for a response when an FDA 483 is issued, and they will review your response. The investigator will get a copy of the FDA 483 response, and the investigator will comment on the response. The compliance office and the investigator enter their comments into a CDRH database. Still, the comments are only general, as to whether the response is adequate or inadequate and will require additional review.
If you do not hear back from the FDA, do not assume that the compliance office or the investigator was satisfied. You should also follow-up several months later (earlier if possible) with a letter that includes evidence of the completed corrective actions, and your verification of effectiveness. If the verification is compelling and received in less than six months of the inspection, you may convince the compliance office to hold off a planned Warning Letter.
If you are interested in root cause analysis and improving your CAPA process, we have two related webinars:
7 Steps to writing an FDA 483 response Read More »