FDA released a final FDA Human Factors Guidance that explains the requirements for human factors testing of medical devices.
FDA Human Factors Guidance
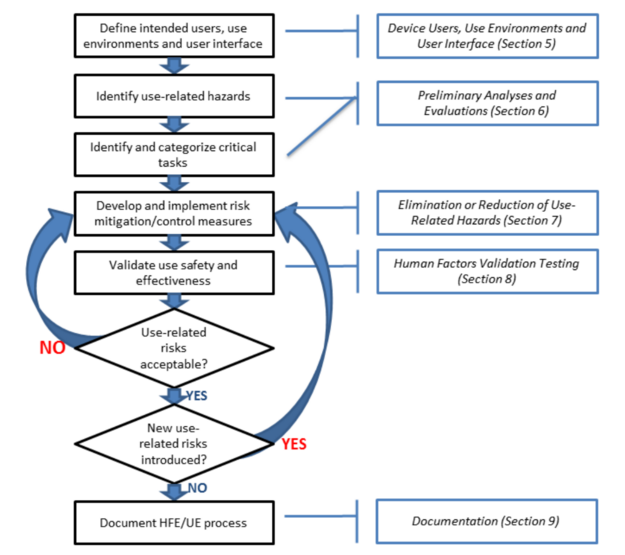
The FDA released a final guidance document on the subject of “Applying Human Factors and Usability Engineering to Medical Devices.” This is a final guidance document that replaces the previous version that was released in 2000 and the draft that was released in 2011. The diagram below is Figure 3 from this new FDA guidance, and it includes references to sections 5 through 9 of the guidance document.

What’s in the FDA human factors guidance?
The organization of the guidance is similar to an ISO standard. Section 1 is the introduction. Section 2 is the scope of the guidance. Section 3 includes definitions, and Section 4 provides an overview of the human factors process and usability engineering as these concepts apply to medical devices. Sections 5 through 9 of the guidance explain the details of the process for applying these concepts to medical devices and risk management. The guidance document includes six references to national and international standards that include human factors engineering or usability engineering, and there are 19 references to articles about human factors engineering and usability engineering at the end of the guidance document.
Incorporating human factors in your design and risk management processes
The steps in the process for human factors engineering and usability engineering mirror the risk management process as defined in ISO 14971 except this guidance does not specify developing a risk management plan or the need to create a risk management file. Although the FDA human factors guidance does not specify that a usability engineering file (UEF) is required, a UEF is required for CE Marking and electrical medical equipment. Therefore, during the design planning phase of your design and development process you should also be identifying the human factors and usability engineering tasks that are needed to complete your UEF. The essential activities to include in your usability engineering plan include:
- creating a use specification (your user needs) – Requirements Phase of Design
- identifying known use errors (design inputs) – Requirements Phase of Design
- performing a task analysis – Development Phase of Design
- creating a use-related risk analysis (URRA) – Development Phase of Design
- conducting formative usability testing – Development Phase of Design
- implementing risk controls – Design “Freeze”
- creating a summative usability protocol – Verification Phase of Design
- writing a summative usability validation report – Validation Phase of Design
Risk controls (i.e., activity #6) are implemented in order to reduce the risk of potential use errors. The unlike other types of risk controls, use-related risk controls require validation instead of verification. The use-related risk controls can be validated through simulated use studies or clinical studies. For these reasons, we recommend documenting use-related risks separately from software risks, security risks, and hardware risks. For IVD 510k submissions, the FDA specifies two different places in the eSTAR to attached your validation documentation. For non-IVD 510k submissions the validation documentation is included in the non-clinical performance testing section of the FDA eSTAR.
Creating a Summative Usability Engineering Report
Clients often ask us what they need to do with regard to human factors engineering and usability engineering for documentation in their technical file (TF) and design history file (DHF). We recommend that they create a summative usability engineering report using the outline provided in the 2016 FDA human factors guidance. We also include a template based on this guidance with our usability procedure (SYS-048) and our human factors training series. If your company makes many types of products with multiple hazard types, then you will need a somewhat generic report template. However, companies with only one or two device families should be able to pre-populate a report template with sections for specific categories of hazards that are applicable to their device family. As discussed in our summative usability report webinar, you can also pre-populate the first seven sections of the report before you conduct summative testing.
Updating Your Risk Management File to Include Usability
As suggested above, we recommend establishing a separate usability engineering file from your risk management file. However, if you already have usability included in your risk management file we recommend updating your risk management file in the following ways:
- update your post-market surveillance plan/risk management plan to specifically gather information about use errors related to the use environment, the user, and the device/user interface
- update your hazard identification report to include hazards related to use errors
- update your risk analysis to include risk controls that you have implemented to reduce the risk of harm related to use errors
- perform and document verification and validation of any new risk controls that you may implement related to use errors
- update your instructions for use to include warnings and precautions about use errors
- develop training tools, such as videos, to demonstrate possible use errors and how to avoid them
The bulk of human factors engineering and usability engineering are documented in the risk management file. Risk management documentation is only required for FDA submissions that include: 1) software, 2) De Novo Classification Requests, and 3) PMA submissions. If you have a non-software device for which you are submitting a 510(k), then you do not include a risk analysis with your submission (exceptions are identified in special controls guidance). It is still critical that design teams address usability engineering, however, because identifying use errors and implementing risk controls to eliminate use errors will prevent product complaints and adverse events. Usability engineering files (UEFs) are also required for electrical medical equipment for IEC 60601-1-6 certification. If these issues are not addressed during the design of a new product, corrective actions and possibly recalls will be needed after the product launch. FDA inspectors will also identify weaknesses in your risk management activities when they identify complaints that are not addressed in your risk analysis.
Additional Training Resources for Usability Engineering
- Human Factors Training Series – $950
- Formative Usability Testing Webinar & Template Bundle – $79
- Use Error and Abnormal Use Decision Tree Training – $79
- Use Specification Template & Webinar Bundle – $79
- Known Use Errors Search Webinar – $129
- Task Analysis Template & Webinar Bundle – $129
- Use-Related Risk Analysis (URRA) Template & Webinar Bundle – $299
- Participant Screening & Data Collection Forms – $79
- Summative Usability Testing Protocol & Webinar Bundle – $199
- Summative Usability Testing Report – Live-streaming Free Webinar
Pingback: FDA Guidance Documents Released Recently -