What are the four types of risk analysis?
You are familiar with design and process risk analysis, but do you know all four types of risk analysis?
Last week’s YouTube live streaming video answered the question, “What are the four different types of risk analysis?” Everyone in the medical device industry is familiar with ISO 14971:2019 as the standard for medical device risk management, but most of us are only familiar with two or three ways to analyze risks. Most people immediately think that this is going to be a tutorial about four different tools for risk management (e.g. FMEA, Fault Tree Analysis, HAZOP, HACCP, etc.). Instead, this article is describing the four different quality system processes that need risk analysis.

What are the four different types?
The one most people are familiar with is risk analysis associated with the design of a medical device. Do you know what the other three are? The second type is process risk management where you document your risk estimation in a process risk analysis. The third type is part of the medical device software development process, specifically a software hazard analysis. Finally, the fourth type is a Use-Related Risk Analysis (URRA) which is part of your usability engineering and human factors testing. Each type of risk analysis requires different information and there are reasons why you should not combine these into one risk management document or template.
Design Risk Analysis
Design risk analysis is the first type of risk analysis we are reviewing in this article. The most common types of design risk analysis are the design failure modes and effects analysis (dFMEA) and the fault-tree analysis (FTA). The dFMEA is referred to as a bottom-up method because you being by identifying all of the possible failure modes for each component of the medical device and you work your way backward to the resulting effects of each failure mode. In contrast, the FTA is a top-down approach, because you begin with the resulting failure and work your way down to each of the potential causes of the failure. The dFMEA is typically preferred by engineers on a development team because they designed each of the components. However, during a complaint investigation, the FTA is preferred, because you will be informed of the alleged failure of the device by the complainant, but you need to investigate the complaint to determine the cause of the failure. Regardless of which risk analysis tool is used for estimating design risks, the risk management process requires that production and post-production risks be monitored. Therefore, the dFMEA or the FTA will need to be reviewed and updated as post-market data is gathered. If a change to the risk analysis is required, it may also be necessary to update the instructions for use to include new warnings or precautions to prevent use errors.
Process Risk Analysis
Process risk analysis is the second type of risk analysis. The purpose of process risk analysis is to minimize the risk of devices being manufactured incorrectly. The most common method of analyzing risks is to use a process failure modes and effects analysis (i.e. pFMEA). This method is referred to as a bottom-up method because you begin by identifying all of the possible failure modes for each manufacturing process step. Next, the effects of the process failure are identified. After you identify the effects of failure for each process step, the severity of harm is estimated. Then the probability of occurrence of harm is estimated, and the ability to detect the failure is estimated. Each of the three estimates (i.e. Severity, Occurrence, and Detectability) are multiplied to calculate a risk priority number (RPN). The resulting RPN is used to prioritize the development of risk controls for each process step.
As risk controls are implemented, the occurrence and detectability scores estimated again. This is usually where people end the pFMEA process, but to complete one cycle of the pFMEA the risk management team should document the verification of the effectiveness of the risk controls implemented. For example, if the step of the process is sterilization then documentation of effectiveness consists of a sterilization validation report. This is the last step of one cycle in the pFMEA, but the risk management process includes monitoring production and post-production risks. Therefore, as new process failures occur the pFMEA is reviewed to determine if any adjustments are needed in the estimates for severity, occurrence, or detectability. If any of the risks increase, then additional risk controls may be necessary. This process is continuously updated with production and post-production information to ensure that process risks remain acceptable.
Software Hazard Analysis
Sofware hazard analysis is becoming more important to medical devices as physical devices are integrated with hospital information systems and with the development of software as a medical device (SaMD). Software risk analysis is typically referred to as hazard analysis because it is unnecessary to estimate the probability of occurrence of harm. Instead, it is only necessary to identify hazards and estimate harm. Examples of these hazards include loss of communication, mix-up of data, loss of data, etc. For guidance on software hazard identification, IEC/TR 80002-1:2009 is a resource. FDA software validation guidance indicates that software failures are systemic in nature and the probability of occurrence cannot be determined using traditional statistical methods. Therefore, the FDA recommends that you assume that the failure will occur and estimate software risks based on the severity of the hazard resulting from the failure.
Use-Related Risk Analysis
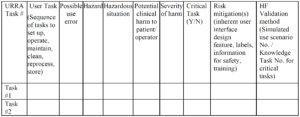 Example of a URRA Table provided by the FDA
Example of a URRA Table provided by the FDACan you use only the IFU to prevent use-related risks?
Facilitating Risk Management Activities – An Interview with Rick Stockton
What are the four types of risk analysis? Read More »