Keeping Design Projects on Schedule: Using the CAPA Process
 The author provides two steps to consider taking now and steps to take in the future that will help keep design projects on schedule using the CAPA process.
The author provides two steps to consider taking now and steps to take in the future that will help keep design projects on schedule using the CAPA process.

The ability to get new, high-quality products to market quickly is the most important core competency for a company to develop. What is your company doing to improve the performance of your design teams? If you have trouble answering the above question, you should consider performing a detailed internal audit of your design control process: http://bit.ly/AuditDesign.
The only significant change I would make to my recommendations from 2012 is to follow the GHTF guidance document for creating technical files using the STED format, instead of using NB-MED 2.5.1/Rec 5: http://bit.ly/GHTFSTEDGuidance. This approach to creating a technical file lends itself to meeting the Canadian Requirements for Medical Device Licensing, and this is the preferred format of Technical Files in Annex II of the proposed EU regulations.
At the end of the blog on how to audit design controls, Step 7 states that you should “Ask the process owner to identify some metrics or quality objectives they are using to monitor and improve the design and development process…If the process owner is tracking no metrics, you might review schedule compliance.” The two most common reasons why design projects are not able to keep pace with the design plan schedule are: 1) there are insufficient resources allocated for the project, and 2) the estimates of the duration for tasks in the schedule are too aggressive.
Step 1: Estimating the Duration of Tasks
Task duration is the easiest quality objective to track performance against. Whether you track design projects with an Excel spreadsheet or Microsoft Project, you can easily compare the actual duration of any project task with the estimated duration that was planned at the beginning of the project. It is important to measure the length of labor hours, rather than using the number of people because most design team members are multi-tasking. You can and should mine the data from previous design projects because this information is available now. As you go through historical data, you will also realize where there are weaknesses in how you capture data regarding labor hours.
Once you have the raw data, I recommend analyzing the data using % deviation and total hours. The % deviation will tell you which estimates were the least accurate, and the total hours will tell you which estimates have the most significant impact on the total project. Ideally, you will collect data from multiple projects, so that you can determine if the deviations are consistent from project-to-project.
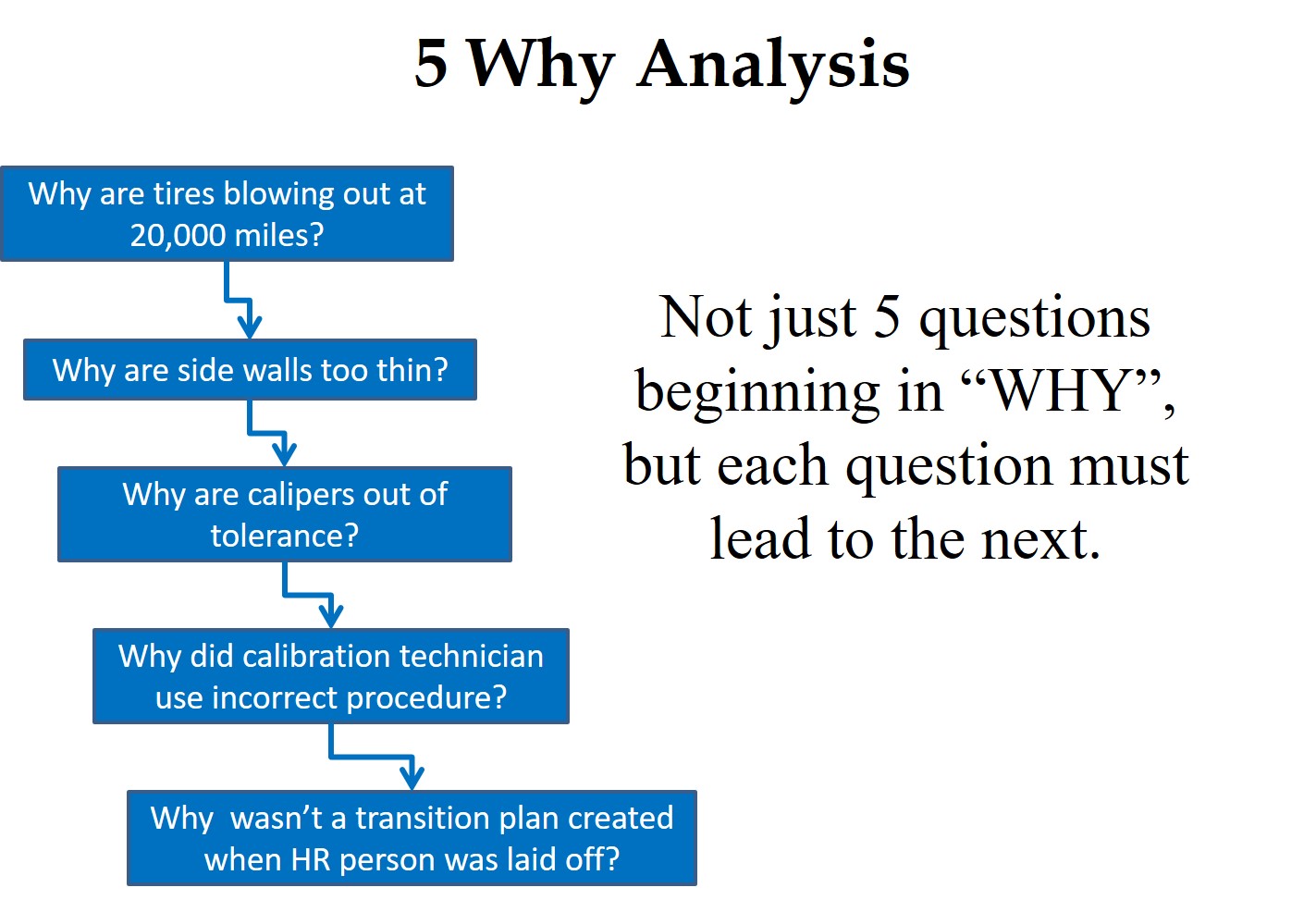
This data analysis is important because the data analysis will help identify the root cause for inaccurate task duration estimates. You may also want to perform a Pareto Analysis of the data to prioritize which project tasks would benefit most from more accurate estimates. Once you have identified the root cause for inaccurate estimates, you can initiate Corrective And Preventive Actions (CAPA), where appropriate.
Step 2: Allocation of Resources
You may hear the phrase “Do more with less,” but I like to joke that design teams are expected to “Do everything with nothing.” If your design team is monitoring the accuracy of taking duration estimates for design projects, the accuracy of your project plans should improve. Your management team should also develop greater confidence in your teams’ ability to forecast product launch dates, thereby the estimates for resource needs. Managers frequently challenge you to achieve the impossible.
The most famous example of this is when Steve Jobs challenged Steve Wozniak to design the video game Breakout in just a few days. If you are the next Steve Jobs, and you have Woz on your team, keep aiming for the moon. If your team consists of mere mortals, you might need more resources. Your senior management may not have additional resources to provide, but it is critical to accurately estimate the resources needed for projects. If you can estimate accurately, you will have the following impact on your company:
- You will be able to estimate the amount of time to market that can be reduced by adding resources.
- You will be able to estimate the cost impact of unrealistic management timelines—instead of saying, “I can’t push it any faster, Captain.” (my favorite Star Trek quote).
Future Steps: Preventive Actions
In one of my previous postings (http://bit.ly/PASources), I stated, “The most fruitful source of preventive actions, however, is data analysis of process control monitoring.” If you are monitoring and measuring your design control process, you can use this approach to:
- Identify preventive actions for your design process
- Establish specifications for critical path tasks in each project
- Calculate your design process capability for successful completion based upon historical data
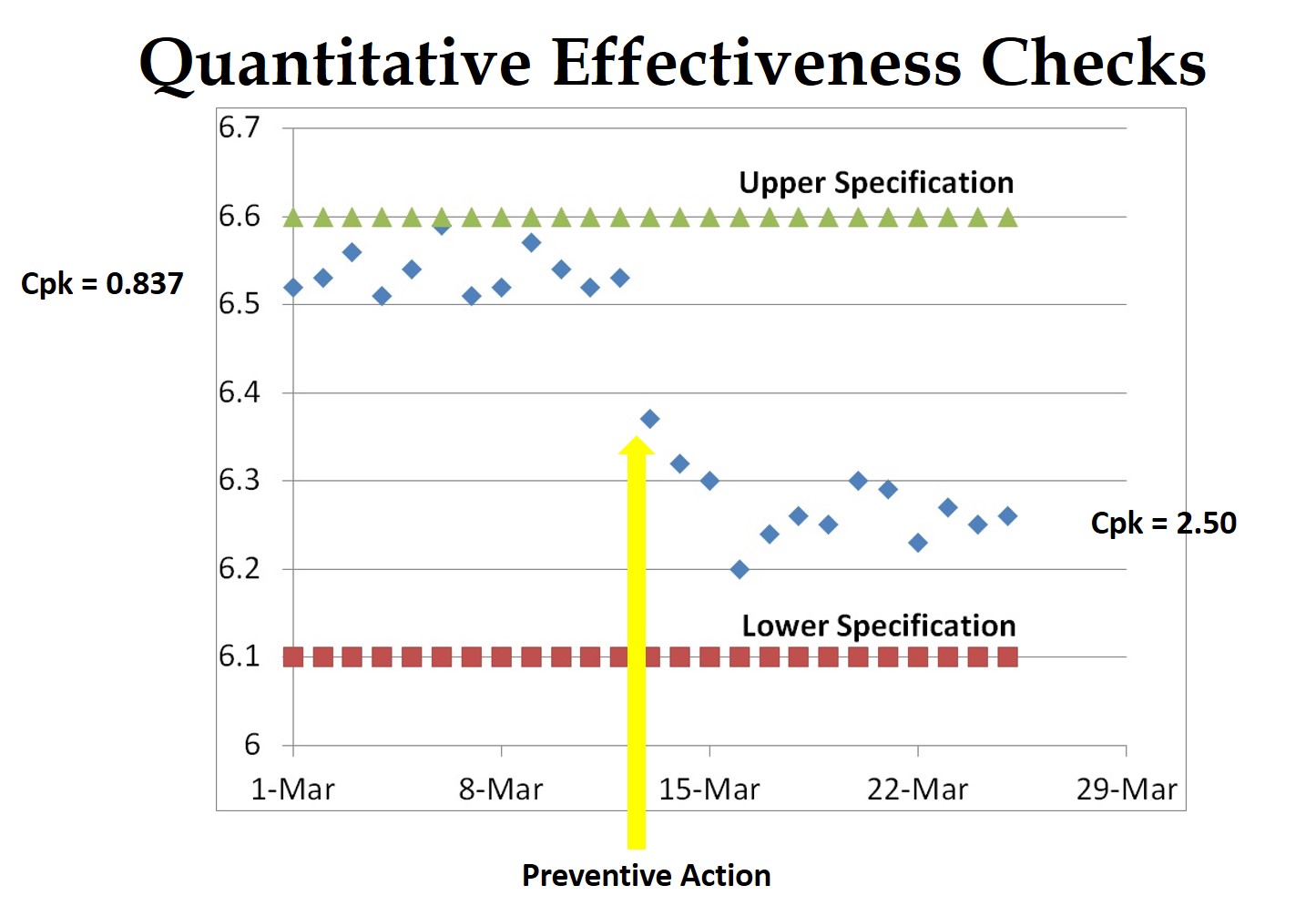
The answer to the following question may help you identify your next design process improvement. How close are your estimates to achieving a Cpk > 1.33 for completing design verification protocols on-time?  If you are interested in learning more about CAPA, please register for the Medical Device Academy’s CAPA Workshop on September 9 in Orlando, or on October 3 in San Diego. Click here to register for the event: http://bit.ly/MDAWorkshops.
If you are interested in learning more about CAPA, please register for the Medical Device Academy’s CAPA Workshop on September 9 in Orlando, or on October 3 in San Diego. Click here to register for the event: http://bit.ly/MDAWorkshops.
Keeping Design Projects on Schedule: Using the CAPA Process Read More »