This will be the 4th edition of our design controls training webinar with new content to modernize your design and development process.
Design Controls Training
Design controls are the #1 cause of FDA 483s. In this design controls training, you will learn how to avoid an FDA 483 inspection observation. Design controls are 1 of the 4 major quality subsystems that the FDA will review during a QSIT level II inspection, and design controls are one of the biggest challenges for companies developing a medical device for the first time. Design controls also apply to any Class 1 device that has software (e.g., MDDS and other software as a medical device or SaMD). This design controls training was recorded on Wednesday, August 14, 2024. It was recorded in two parts (~90 minutes each), and the presentation included 44 slides.
Cost is $129 (AND INCLUDES NATIVE SLIDE POWERPOINT PRESENTATION FILES):
Exam and Training Certificate available for $49.00:
What is covered in design controls training?
Our instructor, Rob Packard of Medical Device Academy, reviews basic concepts and requirements of each design phase of the design process from design plan approval through commercial launch approval, including:
- Design planning
- Design inputs
- Design outputs
- Design verification
- Design validation
- Design reviews and more.
Phase/Gate Approach
You will learn how large companies manage product design-related projects (5-6 hard gates) and keep them on schedule. The lifecycle loop (i.e., post-market surveillance, clinical evaluation, and risk analysis) is illustrated below. This post-market surveillance loop illustrates that design and risk management documentation needs to be updated continuously, and post-market surveillance is an input to the design control process.
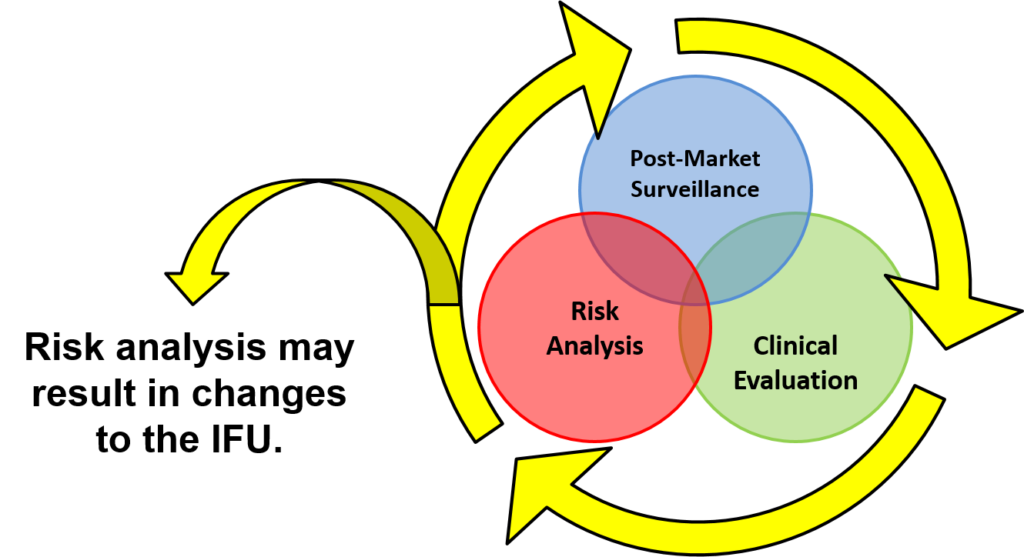
Numerous diagrams are utilized throughout the presentation. Another diagram used in the design controls training shows you how to implement risk management throughout the design process.
The problem with the industry standard approach to the design control process is that it is only meant as a regulatory paperwork exercise. To be an efficient and effective process, design controls need to include documentation that you will actually use. Therefore, in this design controls training, you will learn how to add small details that transform ordinary design documents into development tools that you depend upon.
What are design controls?
Design controls are the process used by medical device manufacturers to ensure that devices and in vitro diagnostics (IVDs) meet the customer and regulatory requirements. Design controls were initially defined in ISO 9001:1994. The ISO process was first adopted by Health Canada and the US FDA, and subsequently integrated into the new ISO 13485 standard in 1996. The design control process consists of seven basic phases:
- Planning
- Identifying Hazards and Requirements
- Development
- Verification
- Validation
- Regulatory Review
- Commercial Release
The design control process adheres to the fundamental principles of the Deming Cycle, also known as PDCA. Phases 1 and 2 are “planning” activities. Phase 3 is the “doing” step in the PDCA cycle. Phases 4, 5, and 6 are “checking” phases. However, you should not begin validation testing (e.g., clinical studies) until you have verified that the current design is safe and effective. You also cannot submit your design for regulatory approval until you have successfully completed all of the verification and validation testing in Phases 4 and 5. Once regulatory approval is received, the company must determine whether the current design is suitable for commercialization. Sometimes, additional design iterations are necessary to enhance safety, performance, usability, or profitability before commercialization.
Planning and project management
Historically, design plans were linear and followed a traditional project management model with a few phases and decision gates:
- Research
- Project approval
- Approval of design inputs
- Approval of design outputs (i.e., “design freeze”)
- Investigational design exemption (IDE) approval (if applicable)
- Regulatory approval
- Commercial release
Unfortunately, designing new products is almost never a linear process. Therefore, most engineers pretend that a new product is still in the “research phase” until the prototype is nearly perfect. As a consequence, the design team doesn’t begin following the design control procedure early enough, and design documentation is started much later than it should be.
Fortunately, lean design and “Agile” methodologies, implemented over the past 25 years, are changing the methods engineers use for design controls. In addition, the medical device industry has adopted international technical standards for risk management (i.e., ISO 14971), software lifecycle management (i.e., IEC 62304), usability engineering (i.e., IEC 62366), and information security management (i.e., ISO 27001). Each of these standards has its own requirement for creating a quality plan. Therefore, today’s modern design planning phase must integrate all four technical disciplines into the medical device design planning process.
Just in case you are wondering which project management model is the best, regulated products require a hybrid model for project management. “Agile” methodologies should be used until the design freeze, and then “Traditional” methodologies should be used after the design freeze. Your design team should also utilize project management software that allows for quick switching between these two models (e.g., Monday.com). The ideal project management software will enable you to utilize both Kanban Boards and Gantt Charts to track project tasks effectively. The perfect software tool will also facilitate communication with internal team members and suppliers outside your company.
Identifying hazards and requirements
Once the design team has created a preliminary design plan, they must next identify the customer and regulatory requirements. Most of these requirements are directly linked to reducing hazards of various types, such as:
- Energy
- Biological
- Chemical
- Performance
- Use-related
As devices and IVDs become more integrated with the Internet of Things (IOT), software hazard analysis and cybersecurity risk assessment have become more important. In fact, for software as a medical device (SaMD), only the software, security, and performance hazards exist.
Iterative development
Once the design team has identified the essential principles for safety and performance, they begin the design and development of the device or IVD. Multiple risk control options are evaluated, and prototypes are created to evaluate the trade-offs of each option being considered. The team will identify and develop test requirements to verify and validate the safety and effectiveness of each risk control. The process is iterative because not every concept works, and the team will discover additional requirements every time a test fails. As the project progresses, the team must update the design plan, document the new requirements, and develop new risk controls.
Design “freeze” and design outputs
Designs are never perfect, but when the team feels the current design is good enough, they will have a “design freeze.” This is when the team switches from an iterative or “Agile” project management model to the traditional project management model (i.e., “Waterfall”). This is not absolutely required, but “freezing” the design is a practical approach because verification and validation testing are the most expensive phase of the design process.
We highly recommend that you conduct a formal design review at the time of your design “freeze” with an independent reviewer. This is not just because an independent reviewer is required for a formal design review; it is also because the independent reviewer will help ensure that your design outputs are thorough and that no verification and validation testing is overlooked in your testing plan. The design outputs are drawings and specifications, while your testing plan consists of a high-level testing schedule and detailed protocols for each test. It is also required to demonstrate traceability of each test back to the original hazards that were identified.
Another element that needs to be considered during your design “freeze” is the status of the other technical documentation. Specifically, your risk management file, software documentation, usability engineering file, and information security management documentation. Each of these four technical disciplines requires that design outputs be documented and that traceability be maintained to the relevant requirements in the testing protocols. The table below provides examples of traceability for each area:
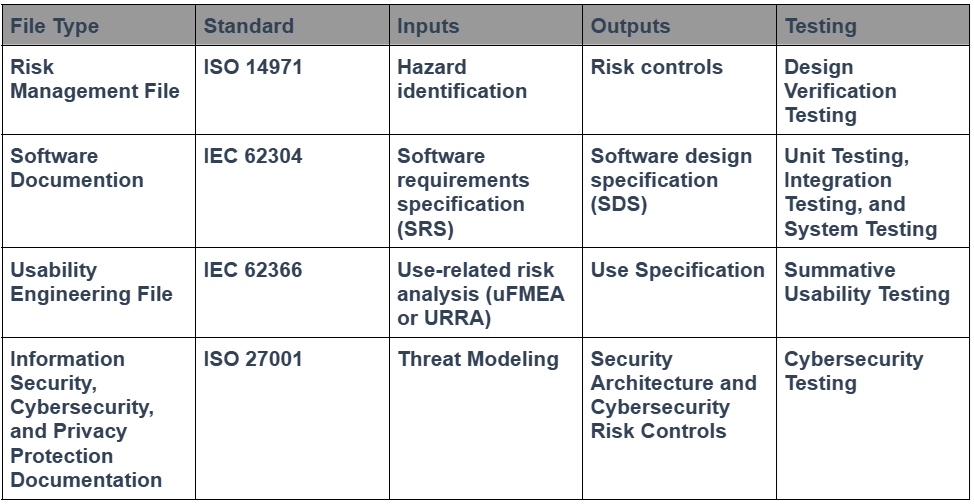
Regulatory experts love tables, and engineers love spreadsheets. We even tried to consolidate all the information you need for a design project in one spreadsheet, but there isn’t enough space in a single spreadsheet cell to include all the necessary details. We now use that spreadsheet solely as a traceability matrix. This is an update to your Design Control Procedure (SYS-008) and the Design Controls Training Webinar. The best tool for documenting traceability, however, is a software database. Therefore, your design team should consider using a modern software database for documenting traceability instead of spreadsheets or tables.
Design verification
Design verification is the process of testing to confirm that the design requirements have been met. Design verification testing is often the most expensive part of the design controls process unless animal studies or human clinical studies are required. The changes we made to the design inputs phase in this design controls training are intended to accelerate design verification progress and minimize errors. Design verification involves creating, reviewing, and approving testing protocols to ensure the accuracy and reliability of the design. You must keep records of the testing results and then generate testing reports. Even if you are outsourcing the verification testing, you still need to review and approve the protocols, and you should review the reports to ensure that the necessary information is included. The FDA will require GLP studies for biocompatibility testing because these tests involve small animal studies. During the webinar, we will emphasize the most recent change in required testing from the FDA, and you will learn which tasks are likely to be critical path items that require advanced planning.
Design validation
Design validation demonstrates that your device can perform the intended use. This typically involves simulated use in a benchtop model, computer analysis (e.g., finite element analysis), cadaver testing, and clinical studies. The human factors process concludes with summative human factors testing. Testing can be considered verification or validation; however, the documentation will be attached as non-clinical benchtop performance testing in the FDA eSTAR. Human factors testing can also be conducted in parallel with human clinical studies using the same subjects. The primary focus of validation testing is demonstrating safety and performance for the intended use, but for FDA 510k submissions, you must also demonstrate equivalence to a predicate device. Equivalence may involve side-by-side comparison testing, and you will need to justify your statistical sample size and acceptance criteria for validation testing because there may not be a recognized standard or guidance specifying these requirements.
Software and cybersecurity updates
The FDA updated software validation requirements on June 14, 2023. The applicable changes impact the classification of software and the risk management documentation required for submission. Additionally, the FDA released its final guidance on cybersecurity on June 27, 2025. The new cybersecurity requirements also impact the design plans for any company that is submitting to the FDA.
Human factors and usability engineering updates
We recorded and updated our usability webinar and released a usability procedure (SYS-048). After carefully listening to the webinar and reading through the usability procedure, we updated our combined design/risk management plan to specify formative testing during iterative development and summative (validation) testing immediately prior to your regulatory submission. You can always modify your own design plans to include this requirement or provide a rationale for why the usability testing does not need to be repeated for device modification. However, my goal was to help explain the value of formative testing and its role relative to summative testing. Minor changes were also made to match the design procedure (SYS-008).
The updated design controls training also includes the impact of the December 2022 draft guidance on Human Factors Engineering. If you are interested in purchasing our usability procedure and template for summative (validation) usability testing, please visit our newest webpage for the usability procedure (SYS-048). Our usability webinar is also available.
It is essential to remember that in the 2016 Guidance Document, “Applying Human Factors and Usability Engineering to Medical Devices,” the FDA notes in the definition of Human Factors Validation Testing that it explicitly represents one portion of the overall design validation of the device.
VIEW OUR PROCEDURES – CLICK HERE OR IMAGE BELOW:

About your design control training instructor

Rob Packard is a regulatory consultant with ~25 years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies, including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.258.1881 or by email. You can also follow him on YouTube, LinkedIn, Facebook, or Instagram.