Effective Management Solutions for 10 CAPA Program Blunders
The author provides effective management solutions for ten real-life CAPA program blunders., (i.e., procedures, root cause analysis, closing CAPAs, etc.)

I am always looking for new and creative ways to help people understand the importance of maintaining an effective Corrective and Preventive Action (CAPA) program. If my last dozen CAPA blogs were not convincing enough, maybe this list of suggestions will help.

- 1. If someone doesn’t follow procedures, just fire them. The employee in question is obviously the root cause. Management cannot be held responsible for the actions of employees. Once, I read a corrective action plan that indicated termination was the correction for a missing training record. The QA Manager clarified this statement by saying that the employee resigned for personal reasons, and there was no opportunity to train the employee. The CAPA record also indicated that 100% of the training records for manufacturing employees were reviewed for completion. There were a few records identified as incomplete, and those employees were trained—not terminated. The company also implemented a tracking tool to monitor training records. As a general rule, termination is not an acceptable corrective action or correction.
- 2. If CAPAs are open longer than your procedure allows, close the existing CAPAs the day before the CAPAs become overdue, and open new CAPAs. CAPAs are not “closed” until all nonconformities have been corrected, corrective and preventive actions are implemented, and effectiveness checks are done. If the corrective and preventive actions were not completely effective, some companies chose to reopen the record and expand the plan of corrective and/or preventive actions. Other companies chose to open a new CAPA record, and reference the new record in the effectiveness check section of the previous CAPA record. Either approach works, but you cannot close an incomplete record and remain compliant.
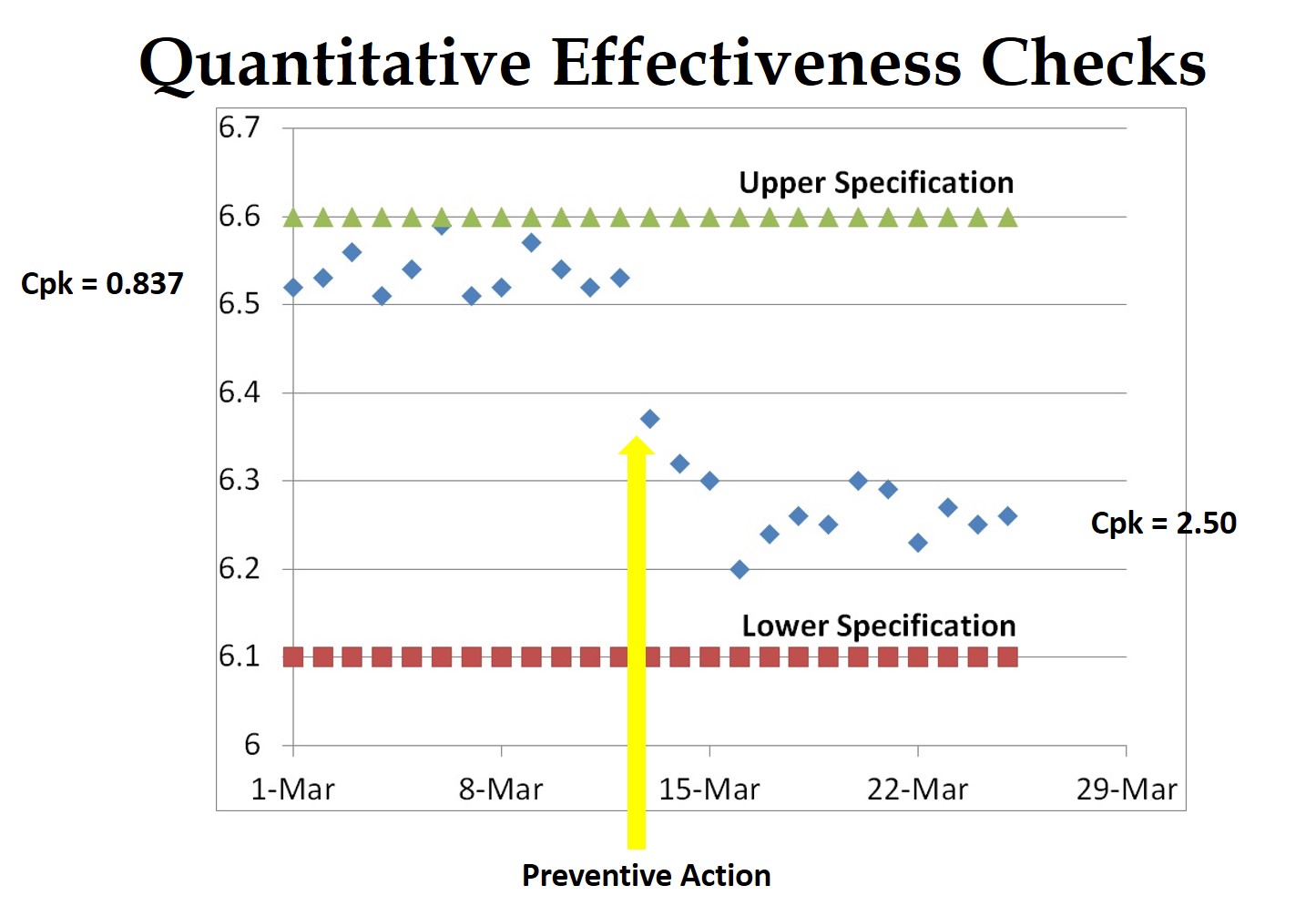
- 3. To verify the effectiveness of corrective actions, just include a copy of your document change order. Documenting changes to procedures meets part of the CAPA requirements, but this verifies implementation—not effectiveness. To verify the efficacy, you need to confirm that a nonconformity, or a potential nonconformity, will not recur. Low-frequency defects are often hard to demonstrate directly. The best approach is to validate the process parameters to demonstrate quantitatively that the process capability has improved. For manual processes, you may need to test the new process to verify that the error will not occur or will be detected.
- 4. If you can’t finish tasks on schedule, revise your plan. If you still can’t finish tasks on schedule, revise your plan again—and again. It’s appropriate to revise your plan if you discover additional causes that your initial investigation missed. You should not, however, be revising target completion dates—except in rare cases. You also should not need to revise your plan multiple times.
- 5. When you’re unsure why a problem occurred, identify the root cause as an unclear procedure, and make a minor change to the appropriate SOP. Making changes to procedures is quick and easy to verify. Unfortunately, this approach is seldom effective in preventing recurrence. You need to develop new process controls to make errors impossible. Eliminate variation in raw materials, eliminate subjectivity in inspections, and provide tools and fixtures to make manual processes capable of more consistent results. After you have reduced all three of these sources for process defects, then you are ready to revise your procedures and retrain employees.
- 6. Whenever an employee fails to follow a procedure, just change the procedure to require another person to verify that they did it right. If one employee fails to follow procedures 100% of the time, a second person manually inspecting will also not be 100% effective. Another method of process control should be used to ensure that your process results in a conforming product. Adding more people provides a false sense of confidence. The use of objective measurement and go/no go fixtures offer a higher degree of certainty.
- 7. Write a justification for an extension of the implementation timeline if a CAPA is about to become overdue. Justifications for extension provide objective evidence that management is aware that a CAPA plan is not meeting the target completion times. This is necessary on rare occasions, but extensions should never become routine. Also, if the progress of a CAPA is slow, monitoring should be frequent enough that management can release additional resources, or re-prioritize assignments in order to catch-up with the target completion date.
- 8. Use the “5 Why” technique for root cause analysis to identify a user error to blame for complaints. The “5 Why” technique is effective at investigating the depth of a problem to ensure that the root cause is identified—instead of a symptom. If the reason for a problem is recognized as a supplier, then it is necessary to ask why the supplier’s error was not prevented or detected. Sometimes this requires asking “Why” more than five times, but identifying a cause you have no control over will fix nothing
- 9. To monitor your CAPA program, conduct weekly CAPA board meetings where a person is asked to explain why the CAPA they were assigned is overdue. Anyone can make an excuse, but excuses will not complete CAPAs. CAPA boards and weekly meetings can be extremely valuable, but your CAPA board should rely on three rules: 1. Managers need to be present to re-allocate resources and re-prioritize tasks. 2. CAPAs that are on schedule or ahead of schedule requires no further discussion. 3. Anyone assigned to a CAPA that is behind schedule should request help and suggest solutions before the CAPA becoming overdue.
- 10. Do not assign other departments the responsibility for CAPAs, because only QA has the training and competency to conduct an investigation of the root cause, and write a CAPA plan. One of the most effective CAPA management tools I observed was a visual communication board that used color-coded paperclips, which identified resources assigned to CAPAs. By limiting the number of paperclips to equal the number of resources allocated to CAPAs, the company was able to level the workload of CAPA assignments to match the available resources in each department. You can only achieve this level of efficiency and effectiveness if multiple people in multiple departments are trained and competent to investigate the root cause and write CAPA plans. CAPA should be a core competency for every department because it’s the best process for fixing and preventing problems.
Disclaimer: If you missed my sarcasm, these are ten ways to mismanage a CAPA program. The brief paragraph after each numbered example is intended to provide the actual recommendation for effective management of your CAPA program.
 If you are interested in learning more about CAPA, please register for the Medical Device Academy’s CAPA Workshop on October 3 in San Diego. Click here to register for the event: http://bit.ly/MDAWorkshops.
If you are interested in learning more about CAPA, please register for the Medical Device Academy’s CAPA Workshop on October 3 in San Diego. Click here to register for the event: http://bit.ly/MDAWorkshops.
Effective Management Solutions for 10 CAPA Program Blunders Read More »