Version 1.0 of the FDA PreSTAR template, released March 29th, now enables the use of the PreSTAR template for 513g requests for information.
What is a 513g request?
A “513g” is a request for classification information from the FDA. The reference is to a Food, Drug & Cosmetic Act section. The purpose of the submission is to ask the FDA what product classification would be most appropriate for your device and what the appropriate regulatory pathway will be. The regulation requires the FDA to provide a written response within 60 days of receiving the 513g request. The submission also requires payment of an FDA user fee eligible for a small business discount.
Is it required to use the new FDA PreSTAR v1.0 template for a 513g request?
No, the FDA PreSTAR is not required to submit a 513g request for information. The FDA has not updated the 2019 guidance document yet, and the FDA continues to allow the use of an FDA eCopy for 513g submissions. However, the updated PreSTAR template simplifies the process of submitting a request for classification information.

What is the required content of a 513g request?
Page 15 of the FDA guidance for 513g requests specifies the following content:
- cover letter,
- description of the device,
- description of what the device is to be used for,
- any proposed labeling or promotional material for the device and, as applicable, any labeling or promotional material of a similar, legally marketed device, if available.
The guidance also details the minimum requirements for these four content requirements. The cover letter requirements specified in the guidance include “your specific question(s) concerning the class in which a device has been classified and/or the regulatory requirements applicable to a device.” When the PreSTAR is used for a pre-submission, there is a designated section at the end of the template for entering questions. However, v1.0 does not allow this option for a 513g. Therefore, questions must be added to the cover letter instead. The template Medical Device Academy created for a 513g includes the following default question:
Reason for the 513(g) Submission:
[Company Name] plans to submit a pre-market submission in 202x, and the company is requesting a decision from the FDA regarding the regulatory pathway for the subject device.
This section can be modified to include additional questions, depending on the specific reason for the 513g request.

When should a 513g request for information be submitted?
Usually, device companies ask me if I think they should submit a 513g or a pre-submission request to answer questions about the testing requirements. Often, the device has a known product classification code that requires a submission of 510(k). Sometimes, there will even be a Special Controls Guidance document available for the product classification. In these situations, a 513g is entirely unnecessary. I can understand the difficulties people experience when navigating the FDA product classification database because the database does not use modern natural language search algorithms like Google. However, a greater concern is that most companies ask this question after they have already started the development of their device and before they plan to initiate design verification testing. This is very late in the design process, and it is even a little late to conduct a pre-submission request. Your 513g submission should be during the beginning of your design project (i.e., during the concept or feasibility phases of design) to verify the proposed regulatory pathway.
How to prepare a 513g
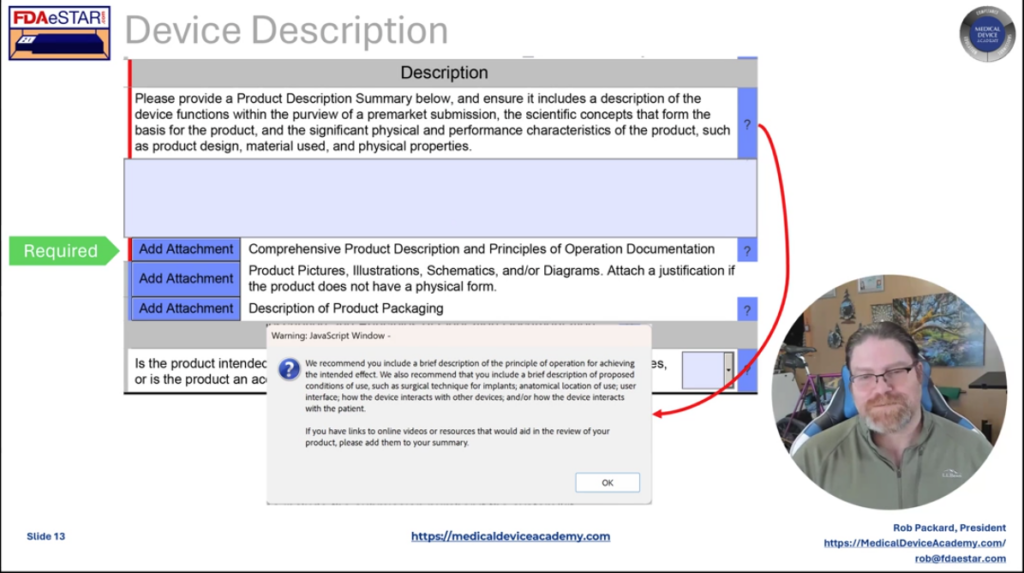
For any device submission, including a 513g, you must prepare a detailed device description for the FDA. Many companies find this difficult. Therefore, we provide a template for the device description. In addition to the device description, we recommend including a copy of the draft labeling and instructions for use (IFU) with each device submission. A pre-submission does not require draft labeling, but a 513g classification request does to ensure the FDA understands your intended use for the device. Therefore, we provide templates for companies to prepare these drafts.
Other Resources
If you need to submit a 513g request, you can learn more about FDA content requirements by watching our 513g submission webinar. You will also receive access to our 513g templates if you purchase the webinar bundle. We also provide the templates for the device description, draft label, and draft instructions for use (IFU) to new clients submitting a pre-submission meeting request, a 510k submission, or a De Novo Classification Request. In addition, there are six (6) other templates included with the 513g webinar bundle. Those templates are specifically required for De Novo submissions, and we recommend including them if you believe your device requires a De Novo submission.
Pingback: Classification Recommendation - How to write one for a De Novo request Medical Device Academy
Since I already have determined the product code (21 CFR 890.3490, Truncal Orthosis, Product Code IQF, Regulation Medical Specialty- Physical Medicine. Product Code IQF called “orthosis, cervical-thoracic, rigid”), is a 513g necessary to be used as a request or clarification? Rather can the form be used as a mechanism to provide the confirmation of the product listing (i.e., 510k exempt and noted as Class I device in accordance with the Product Panel) to FDA instead of asking permission or as a request?
If you are confident of the classification of your device, and you believe it does not exceed the exemption outline in 21 CFR 890.9, then you can list the device with the FDA. You will also need to ensure you meet any other applicable regulations, such as 21 CFR 830, 803, 806, and 820. There is no need to submit a 513(g) or even to fill out a form for review by the FDA. However, if you are not sure of the classification you should review it with a qualified regulatory expert or submit a 513(g) to the FDA.