The 510k course includes 37 new eSTAR webinars, 36+ historical webinars, electronic templates for your next 510k submission, and the eBook “How to Prepare a 510(k) in 100 days.”
The primary advantage of purchasing this 510k Course is that you and your team will receive invitations to new webinars whenever the FDA updates the requirements for a 510k or De Novo submission–at no additional cost. We will also update our existing webinars and templates as we receive feedback from FDA reviewers during the technical screening process, substantive reviews, and interactive reviews. When you purchase our 510k course, you will receive an automated email asking you to confirm your desire to join an email subscription. We deliver all of the updated content through AWeber email subscriptions. If you do not subscribe, you will not receive the updated content. The first email you will receive is to thank you for your purchase and for subscribing to our email updates. The email includes a link to two Dropbox folders (contents as of February 9, 2026):
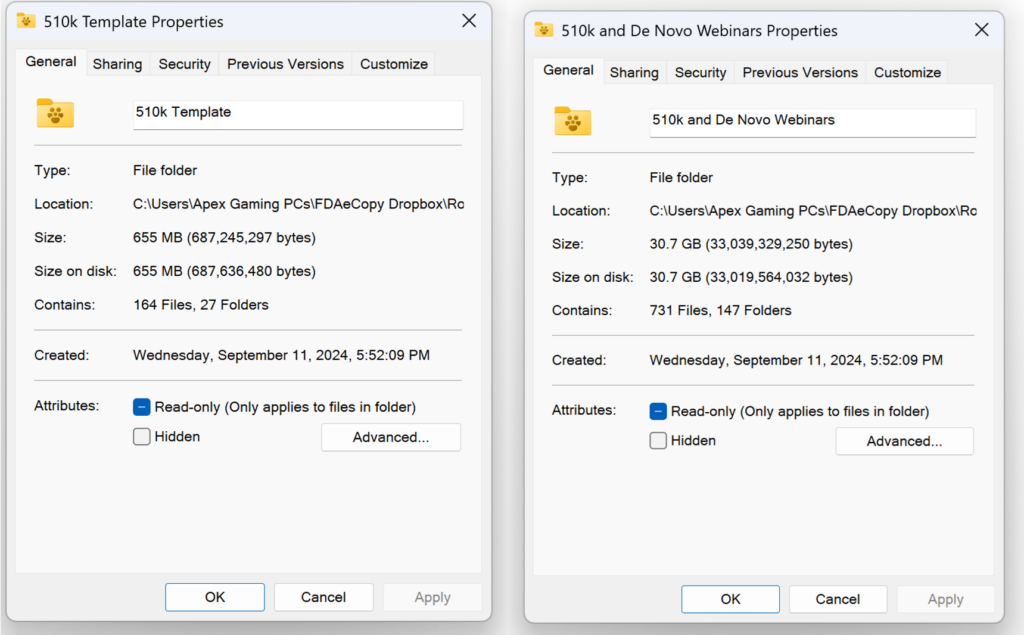
You will need sufficient Dropbox storage space to access the content, but the content can be downloaded and saved locally.
Buy the 510k course for $1,495

Note: If you purchased our new FDA PreSTAR tutorial ($299), you will receive a credit for that prior purchase. Just email Becca Taylor, asking her to email you an invoice for the lower amount, or she can issue a partial refund.
What are the contents of the “510k Template” folder in the 510k Course?
Obsolete 510k templates not intended for use with the FDA eSTAR templates have been archived permanently, and the volume/document structure is no longer used for 510k and De Novo submissions. The screen capture below shows the updated templates folder structure. There are 20 primary templates in the folder–including the two eSTAR templates and three financial disclosure forms provided by the FDA. Most of the content provided in templates is now incorporated into the eSTAR. If you are looking for software or cybersecurity documentation templates, those are sold separately:
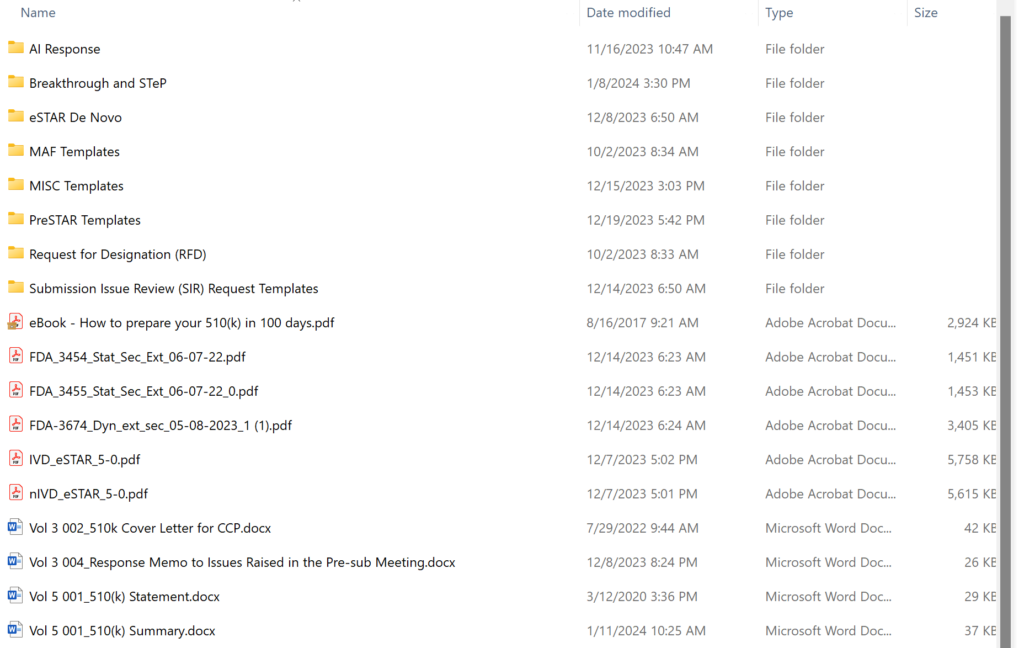
What are the contents of the “510k and De Novo Webinars” folder for the 510k course?
The 510k and De Novo Webinars folder includes all our current and past webinars on 510k submissions. The list below identifies which webinars can be purchased separately and which are only available as part of the 510k Course. The first 33 webinars are listed in the order of the FDA eSTAR from beginning to end.
- FDA eSTAR General Overview – Live-Stream Video
- FDA PreSTAR Tutorial (4-part webinar series) – $299 – Updated for v1.3 (v2.0 only impacts IDE Submissions – see video link below for webinar #36)
- Consensus Standards Webinar – $79 (March 14, 2024)
- Device Description Webinar – Live-Stream, Updated May 9, 2025 @ Noon-1pm ET (Free Template)
- Indications for Use Webinar – $64.50 (August 15, 2024)
- Predicate Selection Webinar – $64.50 (December 8, 2022)
- Special 510(k) Webinar – $79 (March 28, 2024)
- Labeling & UDI 510k Requirements Webinar – $49
- Reprocessing Webinar – $ 79 (October 5, 2024)
- Sterilization Validation Webinar – $79 (October 10, 2024 @ 10:30 a.m. ET)
- Shelf-Life Webinar – $79 (March 21, 2024)
- Biocompatibility Webinar – $79 (February 29, 2024)
- 510k Software Documentation Webinar – Free Download (March 7, 2024)
- Cybersecurity Testing Webinar – Available as part of 510k Course & WI-007
- Interoperability Testing Webinar – $79 (updated August 8, 2024)
- RF Wireless Technology Webinar – Only available as part of 510k Course
- EMC Testing Webinar – Only available as part of 510k Course
- Electrical Safety Testing Webinar – $79 (January 18, 2024)
- Human Factors Webinar – Only available as part of 510k Course
- 510(k) Benchtop Performance Testing Webinar – $79 (January 25, 2024)
- 510(k) Summary – $79 (January 11, 2024)
- Additional Information Request Webinar – Only available as part of 510k Course
- Animal Performance Testing Webinar – $79 (February 1, 2024)
- Human Clinical Study Webinar – $79 (February 8, 2024)
- Dual 510(k) and CLIA Waiver Webinar – $79 (February 15, 2024)
- Test Plan Webinar – $79 (February 22, 2024)
- PreSTAR v1.0 513(g) Template – $79 (April 5, 2024)
- De Novo Risk Mitigation Table – $79 (June 25, 2024)
- Classification Rationale, Regulation Name, and Regulation Number – $49 (July 11, 2024)
- How to document your efforts to identify a potential predicate device? – $79 (July 18, 2024)
- What are your proposed FDA Special Controls for a De Novo? – $79 (July 25, 2024)
- How to prepare a De Novo using an FDA eSTAR (Overview) – $79 (August 1, 2024)
- Benefit-risk analysis webinar – $79 (May 9, 2024)
- What is a pre-ammendment device? – Free (March 14, 2025)
- How to write a request for designation – Free (August 29, 2025)
- What’s new in PreSTAR v2.1? – Only with PreSTAR Tutorial & 510(k) Course (February 10, 2026)
- What’s new in eSTAR v6.1? – Only with 510(k) Course (February 10, 2025)
Other 510k Course Webinars
- Regulatory Submission Plan Webinar – $129 (April 22, 2025)
- 513g Submission Webinar Bundle – $79 (April 5, 2024)
- 510k Submissions: Substantial Equivalence – $129
- FDA eCopy Print & Ship Webinar – Free if you submit a question
- 510k FAQs Webinar – Free if you submit a question
- How to prepare your 510k in 100 days – only available through our 510k Course
- Small Business Qualification Fee for 510(k) Submission – $19
- Redacted FOIA 510k Webinar – $79
- How to Complete a 510k Cover Letter Webinar – $29
- 510k Project Management Best Practices – Free
- 510k Project Management – Updated for 2021 – Free
- In Vitro Diagnostics (IVD) 510k Webinar – $29
- FDA eCopy Print & Ship Webinar – Free if you submit a question
Two new eSTAR templates were released on February 2, 2026:
If you do not submit using v6.1, you may receive additional information requests related to the changes.
- nIVD eSTAR v6.1 (2026-02-02): The PMA QMS section was updated to align with the new Quality Management System Regulation. Minor help text changes were made to the Biocompatibility Attachment G question and citations were changed in multiple places to align with the QMSR. Age groups were added to the Indications for Use section to align with the “Premarket Assessment of Pediatric Medical Devices” guidance. A bug that occurred when modifying the Study Title in the Bench or Clinical attachment questions only after importing data from another eSTAR was fixed. The De Novo alternative practices textbox question in the Device Description section was removed since it was no longer needed. The De Novo classification summary is now requested as an attachment instead of in a textbox.
- Updated 6.0 (2025-10-01): De Novo text now reflects the required use of eSTAR for De Novos. The option to use eSTAR for PMA 30-Day Notice supplements is now enabled. The functionality for Health Canada Additional Information responses was enabled. All the Health Canada help text was enabled. The option to organize attachments by Study Title was added to the Bench and Clinical testing attachments. A button that will delete all deficiency responses was added to the Additional Information section to allow for quick deletion when replying to a second Additional Information request. The biocompatibility color additive help text was updated with additional information.
- Updated 5.6 (2025-07-10): The Cybersecurity section now includes an alternative workflow with fewer questions to account for changes in the updated FDA Cybersecurity guidance. All other IMDRF harmonized attachment questions now use the international help text model (including very minor text changes); Health Canada help text will remain disabled for all sections but Cover Letter and Labeling until approved. A notable number of typo and cosmetic fixes were made. The PMA SSED help text was updated to account for a dead link. Additional guidance information was added to the Clinical Summary textbox help text. FDA Human Factors guidance information was added to the Bench Testing help text. The Sterility packaging test methods help text was updated with examples and references.
- Updated 5.5 (2025-02-12): References to “gender” were removed or changed to “sex.” All harmonized help text in the Administrative Information, Device Description, and Indications for Use sections now uses the international help text model. The Indications for Use section now uses the eSTAR model instead of the Form 3881 format, in preparation for the inclusion of additional jurisdictions. Modifications were made to ensure unacceptable attachment types can’t be added. Certain text boxes (e.g. Labeling) were reformatted to allow more text entry to prevent text cutoff when the eSTAR is printed out to a flat PDF. A modification was made to the validation process to preclude an issue that would have occurred when switching from a complete 30-Day Notice to an incomplete 180-Day Supplement. A bug was fixed that caused a standard deleted from a complete DoC to make the DoC inappropriately incomplete. Minor updates to PCCP AI/ML help text to account for published guidance. The content for PMA 30-Day Notice was added, but the option is disabled until it is approved to deploy. Rich text was reverted to plain text in the Additional Information section due to a bug in PDF-XChange Editor.
- Update 5.4 (2024-11-21): Information was added to the help text of the 510(k) Summary, SSED, Device Description summary, and PCCP question according to PCCP policy clarifications. The Device Description attachment help text for FDA was changed due to clarified Technical Screening criteria and also to be aligned with the international version of the help text. The link provided in the FDA Classification section for a primary product code (e.g., GEI) now goes to the Product Classification website, which was recently updated to include device specific guidance documents. We are evaluating the use of rich text in textboxes (highlight text and right-click for options) by first adding this feature to the deficiency and response textboxes in the Additional Information section (FoxIt isn’t capable of supporting this feature). The ability to reference attachments in eSTAR in the Additional Information section was added below the response textbox. The Standards help text was updated to describe the various ways standards can be added for FDA. The main Cybersecurity help text was updated to clarify alternatives to our recommendations are acceptable with justifications. The Cybersecurity Management Plan help text was modified to refer to the guidance, and the Risks section was removed, since an evaluation of the risks is only needed by the reviewer. The ability to sign the built-in T&A Statement when using FoxIt PDF Reader was disabled due to the tendency for this to corrupt PDFs. eSTAR now asks whether a correspondent or consultant exists in the Applicant Information section with a dropdown (the button was removed).
- Update 5.3 (2024-06-26): The T&A Statement now includes fields for the name and company to ensure they are provided. Multiple minor changes were made based on the upcoming final IMDRF TOC document updates, most notably Software help text updates. The Environment of Use subsection in the Wireless Technology section was removed, since it was redundant with content in the Cybersecurity section.
- Updated 5.2 (2024-03-29): The inclusion of more than 10 standards will no longer cause an Import to run very slowly. An attachment counter was added in the Verification section. A workaround was added to allow the proper display of an eSTAR in Adobe Acrobat Pro when it was prepared with PDF-XChange Editor and formatted text was copied to a textbox. A new international Help Text model was implemented in the Cover Letter and Labeling section to obtain feedback. The wireless function security question was removed from the Wireless Technology section, since it was redundant with content in the Cybersecurity section. AI/ML help text was added to the Software Description attachment question.
- Updated 5.1 (2024-01-08): Hydrogen Peroxide updated as an Established Category A sterilization method, consistent with latest Sterility Guidance.
- Update 5.0 (2023-12-06): PMA content enabled for FDA. Initial international pilot updates (not publicly available yet). EMC Labeling consolidated into a single question, due to typical single citation provided. Guide for Cybersecurity guidance document content added. Pyrogenicity testing options, help text, and EtO residuals question updated in Sterility section. Popup message added if Adobe Acrobat Reader is used. ISO 18562 information added to Biocompatibility section. Cross-section change reminders added to help texts in Device Description section. Clinical Testing section will now display when using PDF-XChange Editor.
- IVD eSTAR v6.1 (2026-02-02): The PMA QMS section was updated to align with the new Quality Management System Regulation (QMSR). Minor help text changes were made to the Biocompatibility Attachment G question and citations were changed in multiple places to align with the QMSR. The title for Indications for Use section was changed to “Intended use/ Indications for Use” and age groups were added to this section to align with the “Premarket Assessment of Pediatric Medical Devices” guidance. Carry-over study and High dose hook effect study were added to Analytical Performance section. The De Novo alternative practices textbox question in the Device Description section was removed since it was no longer needed. The De Novo classification summary is now requested as an attachment instead of in a textbox.
- Updated 6.0 (2025-10-01): De Novo text now reflects the required use of eSTAR for De Novos. The option to use eSTAR for PMA 30-Day Notice supplements is now enabled. Stability related requests are relocated from Performance Testing section to Shelf-Life and Stability section. Health Canada help texts in many sections were enabled. The functionality for Health Canada Additional Information responses was enabled. A button that will delete all deficiency responses was added to the Additional Information section to allow for quick deletion when replying to a second Additional Information request. The biocompatibility color additive help text was updated with additional information.
- Update 5.6 (2025-07-10): The Cybersecurity section now includes an alternative workflow with fewer questions to account for changes in the updated FDA Cybersecurity guidance. Many IMDRF harmonized attachment questions now use the international help text model (including very minor text changes); Health Canada help text will remain disabled for all sections but Cover Letter and Labeling until approved. A notable number of typo and cosmetic fixes were made. The PMA SSED help text was updated to account for a dead link. Additional guidance information was added to the Clinical Summary textbox help text. The Sterility packaging test methods help text was updated with examples and references.
- Update 5.5 (2025-02-12): References to “gender” were removed or changed to “sex.” All harmonized help text in the Administrative Information, Device Description, and Indications for Use sections now uses the international help text model. The Indications for Use section now uses the eSTAR model instead of the Form 3881 format, in preparation for the inclusion of additional jurisdictions. Modifications were made to ensure unacceptable attachment types can’t be added. Certain text boxes (e.g. Labeling) were reformatted to allow more text entry to prevent text cutoff when the eSTAR is printed out to a flat PDF. A modification was made to the validation process to preclude an issue that would have occurred when switching from a complete 30-Day Notice to an incomplete 180-Day Supplement. A bug was fixed that caused a standard deleted from a complete DoC to make the DoC inappropriately incomplete. Minor updates to PCCP AI/ML help text to account for published guidance. The content for PMA 30-Day Notice was added, but the option is disabled until it is approved to deploy. Rich text was reverted to plain text in the Additional Information section due to a bug in PDF-XChange Editor.
- Update 5.4 (2024-11-21): Information was added to the help text of the 510(k) Summary, SSED, Device Description summary, and PCCP question according to PCCP policy clarifications. The Device Description attachment help text for FDA was changed due to clarified Technical Screening criteria and also to be aligned with the international version of the help text. The link provided in the FDA Classification section for a primary product code (e.g., GEI) now goes to the Product Classification website, which was recently updated to include device specific guidance documents. We are evaluating the use of rich text in textboxes (highlight text and right-click for options) by first adding this feature to the deficiency and response textboxes in the Additional Information section (FoxIt isn’t capable of supporting this feature). The ability to reference attachments in eSTAR in the Additional Information section was added below the response textbox. The Standards help text was updated to describe the various ways standards can be added for FDA. The main Cybersecurity help text was updated to clarify alternatives to our recommendations are acceptable with justifications. The Cybersecurity Management Plan help text was modified to refer to the guidance, and the Risks section was removed, since an evaluation of the risks is only needed by the reviewer. The ability to sign the built-in T&A Statement when using FoxIt PDF Reader was disabled due to the tendency for this to corrupt PDFs. eSTAR now asks whether a correspondent or consultant exists in the Applicant Information section with a dropdown (the button was removed).
- Update 5.3 (2024-06-26): The T&A Statement now includes fields for the name and company to ensure they are provided. Multiple minor changes were made based on the upcoming final IMDRF TOC document updates, most notably Software help text updates. The Environment of Use subsection in the Wireless Technology section was removed, since it was redundant with content in the Cybersecurity section.
- Update 5.2 (2024-03-29): The inclusion of more than 10 standards will no longer cause an Import to run very slowly. An attachment counter was added in the Verification section. A workaround was added to allow the proper display of an eSTAR in Adobe Acrobat Pro when it was prepared with PDF-XChange Editor and formatted text was copied to a textbox. A new international Help Text model was implemented in the Cover Letter and Labeling sections to obtain feedback. The wireless function security question was removed from the Wireless Technology section, since it was redundant with content in the Cybersecurity section. AI/ML help text was added to the Software Description attachment question.
- Update 5.1 (2024-01-08): Hydrogen Peroxide updated as an Established Category A sterilization method, consistent with updated Sterility Guidance.
- Update 5.0 (2023-12-06): PMA content enabled for FDA. Initial International pilot updated (not publicly available yet). EMC Labeling consolidated into a single question, due to typical single citation provided. Guide for Cybersecurity guidance document content added. Pyrogenicity testing options, help text, and EtO residuals question updated in Sterility section. popup message added if Adobe Acrobat Reader is used. ISO 18562 information added to Biocompatibility section. cross-section change reminders added to Help Texts in Device Description section.
A new PreSTAR template was released on February 2, 2026:
If you do not submit using v2.0, you may receive additional information requests related to the changes.
- PreSTAR v2.1 (2025-10-01): Updated help text to match the new QMSR regulations references. Added pediatric population checkboxes in intended use section to align with “Premarket Assessment of Pediatric Medical Devices” guidance. Typographic and minor logic updates to further align IDE with other eSTAR and internal reviews and to inform IDE submitters that certain eSTAR sections can be disabled in the device description section if they are not applicable to an IDE supplement review.
- Update 2.0 (2025-10-01): Templates for IDE Originals and Supplements, as well as their amendments, were integrated into the eSTAR. Minor general modifications were made to the Product Listing in the Device Description, Indications for Use, and Other Labeling in the Labeling subforms to account for the integration of IDE.
- Update 1.4 (2025-07-10): Updated to be consistent with the nIVD and IVD eSTARs. A notable number of typo and cosmetic fixes were made.
- Update 1.3 (2025-02-12): References to “gender” were removed or changed to “sex.” Modifications were made to ensure unacceptable attachment types can’t be added. Certain text boxes (e.g. Labeling) were reformatted to allow more text entry to prevent text cutoff when the eSTAR is printed out to a flat PDF. Minor updates to PCCP AI/ML help text to account for published guidance. List of Standards was updated. Rich text was reverted to plain text in the Additional Information section due to a bug in PDF-XChange Editor. Minor bugs and typos were fixed.
- Update 1.2 (2024-11-21): Information was added to the help text of the Product Description summary and PCCP question according to PCCP policy changes. The link provided in the FDA Classification section for a primary product code (e.g., GEI) now goes to the Product Classification website, which was recently updated to include device specific guidance documents. We are evaluating the use of rich text in textboxes (highlight text and right-click for options) by first adding this feature to the deficiency and response textboxes in the Additional Information section (FoxIt isn’t capable of supporting this feature). The ability to reference attachments in eSTAR in the Additional Information section was added below the response textbox. The Standards help text was updated to describe the various ways standards can be added for FDA. eSTAR now asks whether a correspondent or consultant exists in the Applicant Information section with a dropdown (the button was removed). The Device Description attachment help text for FDA was changed to be consistent with the nIVD and IVD eSTARs.
- Update 1.1 (2024-06-26): Modified the categorization of Application/Submission types. Updated list of Standards. Updated FAQ. Fixed minor bugs and typos.
- Update 1.0 (2024-03-29) from version 0.3: 513(g) submission content was enabled. The inclusion of more than 10 standards will no longer cause an Import to run very slowly. An attachment counter was added in the Verification section. A workaround was added to allow the proper display of an eSTAR in Adobe Acrobat Pro when it was prepared with PDF-XChange Editor and formatted text was copied to a textbox. The FAQ was updated. The list of standards, list of regulations, and list of product codes were updated. Other minor bugs were fixed and minor changes were implemented.
- Update 0.3 (2023-12-06) from version 0.2: Standard recognition number can now be used to autopopulate standard information. Updated Software guidance document link. Updated list of medical device product codes. Updated FAQ. Fixed minor bugs.
- Update 0.2 (2023-08-14) from version 0.1: Addition of PCCP submission topic category. Minor text changes.
- Updated 0.1 (2023-06-06): Initial beta release.
Without knowing what design stage you are in, we can’t tell you where to start watching our 510k course webinars. However, the following YouTube video might help you figure out where you are in the design process. The video has timestamps that allow you to advance and rewatch any video section. Many start-up companies try to begin the 510k process without a design plan. They intend to create the design plan and design history file (DHF) retrospectively after they submit their 510k submission. However, this is a mistake. You should always start your 510k project with a design plan. The primary reason is that it will help you create a realistic timeline for the overall project. The second most important reason for starting with design planning is that it will force you to schedule the testing required for verification and validation (V&V) testing and any pre-requisites that must be completed before you begin V&V testing. We have another form allowing you to download our design plan template and 510k table of contents. These two documents are the primary planning tools we recommend you use to plan your 510k project.
To give you an idea of what we have included in our 510k course, below is a YouTube video recorded with Joe Hage in October 2018 explaining how to prepare a 510k submission.
Dear Rob,
I would like to ask for a quotation for the 510(k) courses and a list of the content included.
We recently start working with a US 510(k) project, and I think it is good to get my knowledge on 510(k) up to date. I have attended a few of your free webinars and a RAPS SaMD training, very helpful to my professional development as a RA specialist.
I also have the question of update plan from your side, as FDA published quite a few guidance recently. If I purchase the course now, will I get free access to the update webinars and updated templates?
Thanks a lot.
May
Dear May,
Thank you for your inquiry. If anyone has trouble with purchases on our website, needs a quote, or a receipt is needed; you can request a quote, invoice, or receipt directly from Becca Taylor (becca@medicaldeviceacademy.com) or Lindsey Walker (sales@medicaldeviceacademy.com). I will personally send you an invoice for the 510(k) course in a few moments, and you are welcome to contact me directly to ask any technical questions.
Regarding updates – I already conducted a webinar this week about the predicate selection draft guidance. Another webinar is planned for next week on biocompatibility, but I have not updated the website yet. If you purchase the course now, you get invites to updated webinars and access to all of the recording in the future. Any new templates we create you will also have access to, but the number of templates is decreasing with the implementation of the eSTAR.
Rob
Hello Rob!
I would like to know if you have some templates and explanation of what the FDA expects for an SaMD with deep learning in a 510k process?
Thanks in advance for your reply.
Have a nice day.
For any SaMD product the FDA expects details in the device description. For AI and ML products, details about the algorithm must be included as well. Our device description template includes information about this.
Does a products design and process validation need to be completed before a 510(k) is submitted to the FDA?
Very good question. There are many opinions, but I will be specific to legal requirements. The 510(k) requires that you submit designs and specifications in the device description section, draft labeling in the labeling section, and V&V testing reports in the other sections of the eSTAR. Therefore, all of your design verification and validation must be completed in order to even complete the eSTAR. Process validation is required for certain processes, such as sterilization and packaging. However, it is common for companies to scale-up a process during and after a 510(k). Therefore, additional process validation and possibly some design verification might need to be repeated. The design reviews, design plan, and all of your design transfer activities are not required in a 510(k) submission, but these documents are required for compliance with the QSR and ISO 13485. These other items would be sampled during inspections and/or audits. Your team should not be agreeing to release a product commercially until the DHF is complete and the DMR is approved.
Dear Rob,
I’m very interested in learning about FDA 510K submissions, however, this is my first time dealing with this process. My experience lies in submitting to health agencies in Latin America, and your free content has helped me understand much more. However, I’d like to continue learning. Which programs from your course do you recommend for getting started and completing a 510K submission?
Thanks, have a nice day!
I recommend starting with video #1 and working your way down the list. When you are finally ready to purchase the course as a whole, please contact Lindsey Walker to upgrade to the full course. She will credit you for previous purchases of the individual webinars. If the devices you work on are similar in nature, you might be able to skip certain webinars that cover testing that is not applicable to your devices. For example, biocompatibility is not applicable to SaMD products.