Software validation documentation for a medical device
Learn why you need to start with software validation documentation before you jump into software development.
When do you create software validation documentation for a medical device or IVD?
At least once a week, I speak with the founder of a new MedTech company that developed a new software application as a medical device (SaMD). The founder will ask me to explain the process for obtaining a 510(k), and they want help with software validation documentation. Many people I speak with have never even heard of IEC 62304.

What are the 11 software validation documents required by the FDA?
In 2005 the FDA released a guidance document outlining software validation documentation content required for a premarket submission. There were 11 documents identified in that guidance:

What the FDA guidance fails to explain is that some of these documents need to be created before software development begins, or your software validation documentation will be missing critical design elements. Therefore, it is important to create a software development plan that schedules activities that result in those documents at the right time. In contrast, four of the eleven documents can wait until your software development is complete.
Which of the software validation documents can wait until the end?
The level of concern only determines what documents the FDA wants to review in a submission rather than what documents are needed for a design history file. In fact, the level of concern (LOC) document is no longer required as a separate document in premarket submissions using the FDA eSTAR template because the template already incorporates the questions that document your LOC. The revision level history document is simply a summary of revisions made to the software during the development process, and that document can be created manually or automatically at the end of the process, or the revision level history can be a living document that is created as changes are made. The traceability matrix can also be a living document created as changes are made, but its only purpose is to act as a tool to provide traceability from hazards to software requirements, to design specifications, and finally to verification and validation reports. Other software tools, such as Application Lifecycle Management (ALM) Software, are designed to ensure the traceability of every hazard and requirement throughout the entire development process. Finally, unresolved anomalies should only be documented at the time of submission. The list may be incomplete until all verification and validation testing is completed, and the list should be the shortest at the time of submission.
What documentation will be created near the end of development?
The software design specification (SDS) is typically a living document until your development process is completed, and you may need to update the SDS after the initial software release to add new features, maintain interoperability with software accessories, or change security controls. The SDS can not begin, however, until you have software requirements and the basic architecture defined. The verification and validation activities are discrete documents created after each revision of the SDS and must therefore be one of the last documents created–especially when provided to the FDA as a summary of the verification and validation efforts.
Which validation documents do you need first?
At the beginning of software development, you need a procedure(s) that defines your software development process. That procedure should have a section that explains the software development environment–including how patches and upgrades will be controlled and released. If you don’t have a quality system procedure that defines your development process, then each developer may document their coding and validation activities differently. That does not mean that you can’t improve or change the procedure once development has begun, but we recommend limiting the implementation of a revised procedure when making major software changes and discussing how revisions will be implemented for any work that remains in progress or has already been completed.
When do the remaining software validation documents get created?
The remaining four software validation documents required for a premarket submission to the FDA are:
- Software description
- Software hazard analysis
- Software requirements specification (SRS)
- Architecture design chart
Your development process will be iterative, and therefore, you should be building and refining these four documents iteratively in parallel with your software code. At the beginning of your project, your design plan will need a brief software description. Your initial software description needs to include the indications for use, a list of the software’s functional elements, and the elements of your user specification (i.e., intended patient population, intended users, and user interface). If you are using lean startup methodology, the first version of your device description will be limited to a minimal viable product (MVP). The target performance of the MVP should be documented as an initial software requirements specification (SRS). This initial SRS might only consist of one requirement, but the SRS will expand quickly. Next, you need to perform an initial software hazard analysis to identify the possible hazards. It is important to remember that software hazards are typically hazardous situations and are not limited to direct physical harm. For each potential hazard you identify in your hazard analysis, you will need a software requirement to address each hazard, and each requirement needs to be added to your SRS. As your software becomes more complex by adding software features, your device description needs to be updated. As you add functions and requirements to your software application, your SRS will need updates too. Finally, your development team will need a tool to track data flow and calculations from one software function to the next. That tool is your architecture design chart, and you will want to organize your SRS to match the various software modules identified in your architecture diagram. This phase is iterative and non-linear, you will always have failures, and typically a team of developers will collaborate virtually. Maintaining a current version of the four software documents is critical to keeping your development team on track.
How do you perform a software hazard analysis?
One of the most important pre-requisite tasks for software developers is conducting a hazard analysis. You can develop an algorithm before you write any code, but if you start developing your application to execute an algorithm before you perform a software hazard analysis, you will be missing critical software requirements. Software hazard analysis is different from traditional device hazard analysis because software hazards are unique to software. A traditional device hazard analysis consists of three steps: 1) answering the 37 questions in Annex A of ISO/TR 24971:2020, 2) systematically identifying hazards by using Table C1 in Annex C of ISO 14971:2019, and 3) reviewing the risks associated with previous versions of the device and similar competitor devices. A software hazard analysis will have very few hazards identified from steps 1 and 2 above. Instead, the best resource for software hazard analysis is IEC/TR 80002-1:2009. You should still use the other two standards, especially if you are developing software in a medical device (SiMD) or firmware, but IEC/TR 80002-1 has a wealth of tables that can be used to populate your initial hazards analysis and to update your hazard analysis when you add new features.
How do you document your hazard analysis?
Another key difference between a traditional hazard analysis and a software hazard analysis is how you document the hazards. Most devices use a design FMEA (dFMEA) to document hazards. The dFMEA is a bottom-up method for documenting your risk analysis by starting with device failure modes. Another tool for documenting hazards is a fault tree diagram.
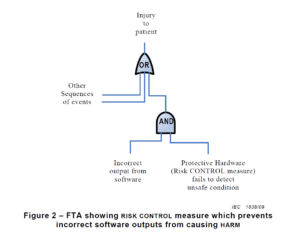
A fault tree is a top-down method for documenting your risk analysis, where you identify all of the potential causes that contribute to a specific failure mode. Fault tree diagrams lend themselves to complaint investigations because complaint investigations begin with the identification of the failure (i.e., complaint) at the top of the diagram. For software, the FDA will not allow you to use the probability of occurrence to estimate risks. Instead, software risk estimation should be limited to the severity of the potential harm. Therefore, a fault tree diagram is generally a better tool for documenting software risk analysis and organizing your list of hazards. You might even consider creating a separate fault tree diagram for each module of your software identified in the architecture diagram. This approach will also help you identify the potential impact of any software hazard by looking at the failure at the top of the fault tree. The higher the potential severity of the software failure, the more resources the software team needs to apply to developing software risk controls and verifying risk control effectiveness for the associated fault tree.
Software validation documentation for a medical device Read More »