The format and content requirements for an FDA pre-submission have not changed, but the launch of the FDA PreSTAR has changed everything.
What is an FDA pre-submission?
An FDA pre-submission aims to get answers to questions you have about a future FDA submission. The pre-submission may consist of one large PDF document or multiple PDF documents. In your pre-submission, you must select either an email response or an email response with a teleconference. One advantage of choosing a teleconference is that you can ask clarifying questions during a one-hour teleconference with the FDA. Still, you are responsible for submitting draft meeting minutes to the FDA within 15 days of the teleconference. If you select an email response, you do not need to provide meeting minutes to the FDA.

Our new 4-part FDA pre-submission webinar series is available on-demand. You can download it and watch it as many times as you want. This will be the Ultimate FDA pre-submission training. Do not miss it.

Everyone asks us for examples, so we will show you how to complete the entire FDA PreSTAR for a device in this webinar series. If you would like to vote on which device we should use as an example (i.e., Option 1 = Infrared Thermometer or Option 2 = Antimicrobial Gauze), please place your vote on our LinkedIn page.
What is the difference between an FDA pre-submission and a Q-submission?
Every FDA pre-submission is a Q-submission, but not all Q-submissions are pre-submissions. The new PreSTAR template is currently limited to an FDA pre-submission, but the template will be expanded to other types of Q-subs later. The FDA pre-submission template (i.e., PreSTAR) beta version 0.1 is unnecessary for responses to interactive review questions from the FDA. Just email the Lead Reviewer (file size limit is 25 MB for email).
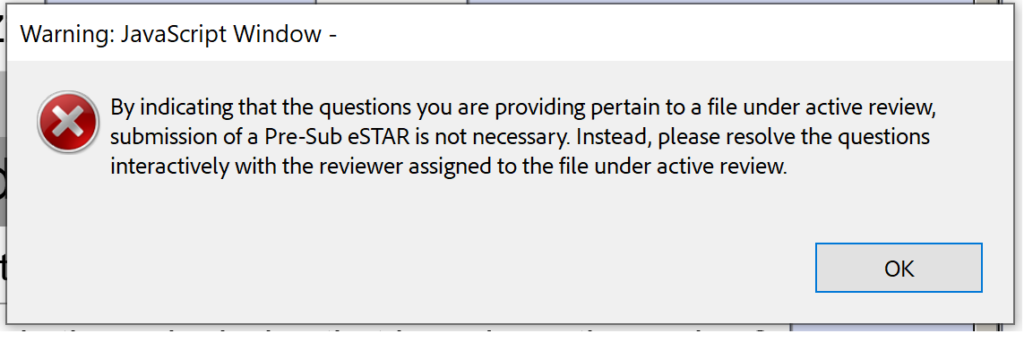
Unfortunately, the beta version 0.1 is also not ready for Submission-in-Review (SIR) meetings or responses to IDE during an interactive review.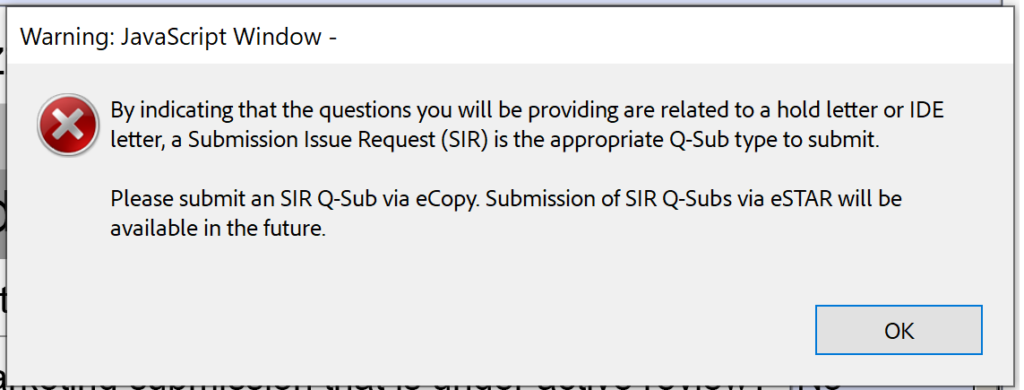
13 other types of submissions might benefit from Q-submissions:
- Submission Issue Requests (SIRs)
- Study Risk Determinations
- Informational Meetings
- Breakthrough Device Designation Requests
- Informational Meetings
- PMA Day 100 meetings
- Agreement and Determination meetings
- Submissions associated with the STeP program
- Accessory classification requests
- Requests for FDA feedback on specific questions or cross-cutting policy matters
- Requests for recognition of publicly accessible genetic variant databases
- Combination product agreement meetings (CPAM), and
- Feedback on FDA 483 inspection observations.
We expect the PreSTAR template to eventually be available for a 513(g) request in the future because it was already validated for that purpose.
What is the Q-submission number?
All Q-submissions are assigned a document number beginning with “Q” upon receipt (i.e., Qyyxxxx). The format of the number consists of 2-digits (i.e., “yy”) for the year of submission (e.g., “23” for 2023) and 4-digits (i.e., “xxxx”) that are the following sequential number assigned by the FDA for that calendar year. Therefore, the first Q-submission received by the FDA in January 2023 is Q230001, and between 3,500 and 4,000 new submissions are usually received each year. If the subject device was submitted in a previous Q-submission, the original document number is re-used, and a supplement number is added (i.e., Qyyxxx/S001, Qyyxxx/S002, etc.). Q-submission numbering is explained in more detail in the 2023 FDA guidance.
Does the FDA charge for Q-submissions?
FDA pre-submissions do not require paying an FDA User Fee (i.e., $0).
How long does an FDA pre-submission take?
The days of squishing timelines are gone. The timeline is 70-75 calendar days. On October 5, 2022, MDUFA V was approved. As one of the MDUFA V decision targets, the FDA is tasked with reducing the timeline for responding to pre-sub questions within 70 days for 90% of pre-sub requests. The FDA is tasked with achieving this goal by March 2024. If they are successful, the FDA will receive an increase of 59 headcounts to their budget in 2024. This is approximately a $19 million incentive to respond to your pre-submission meeting questions within 70 days. To reflect these new MDUFA V decision targets, the FDA updated the Q-Sub guidance document to reflect the target date of 70 days for the email response and 75 days for teleconference meetings. The FDA also updated the Customer Collaboration Portal (CCP) to facilitate tracking of FDA pre-submission deadlines.
What is an FDA PreSTAR?
In the past, you had to create your document(s) for an FDA pre-submission. Some people create one large PDF document divided into sections, while others create separate PDF documents for each requirement of the FDA pre-submission guidance. On August 14, 2023, the FDA released an updated beta version (i.e., version 0.2) of a new PDF template (i.e., FDA PreSTAR). This new PreSTAR template provides multiple benefits to the FDA:
- every company uses the same format,
- the template automatically verifies that the pre-submission includes all required elements, and
- Including optional elements will encourage companies to provide more device details than they might otherwise provide.
The PreSTAR also benefits submitters:
- you will never forget the required elements of the FDA pre-submission,
- you never have to validate an FDA eCopy, and
- the similar format and user interface will train you to use the FDA eSTAR.
Note: October 1, 2023, was the FDA eSTAR implementation deadline.
Do you have to use the PreSTAR template?
Nope. The PreSTAR version 0.2 is a beta version and 100% optional. However, I like it better than my templates. Your design team can still have individual documents for the user manual, device description, and testing plan. We attach the document using the button that says “Add Attachment” (see screen capture below).

The PreSTAR template was built by Patrick Macatangga, a Tools & Templates Engineer working at the FDA on the Lifecycle Tools and Templates Team. To help with where to direct questions about the template, he suggested:
- If you have questions or feedback regarding the voluntary use of the eSTAR for medical devices regulated by CDRH, or if you have general questions about medical devices, contact the Division of Industry and Consumer Education (DICE).
- If you find malfunctions or errors in the eSTAR template for medical devices regulated by CDRH, contact eSubPilot@fda.hhs.gov.
- If you have questions regarding 510(k)s, De Novo requests, or Early Submission Requests for medical devices regulated by CDRH, contact OPEQSubmissionSupport@fda.hhs.gov.
How do you submit an eCopy?
You can submit an FDA eCopy on electronic media (e.g., USB flash drive) and send it via FedEx to the FDA Document Center at the following address: Food and Drug Administration, Center for Devices and Radiological Health, Document Mail Center, 10903 New Hampshire Ave., Bldg. 66, rm. G609, Silver Spring, MD 20993-0002. However, you can also submit an FDA eCopy via a web browser (i.e., CCP…see next section on submitting a PreSTAR).
If you are submitting an eCopy through the CCP instead of an FDA PreSTAR
How do you submit a PreSTAR?
You have two options for delivery of an FDA pre-submission:
- save the pre-sub on electronic media (e.g., USB flash drive) and send it via FedEx to the FDA, and
- upload the pre-sub to the new FDA Customer Collaboration Portal (CCP).
As you can guess from the video above, we only use option 2 for FDA pre-submissions. For option 2, you can upload an eCopy (saved as a zip file) or a PreSTAR (in the native PDF format). The image below shows you how this is done, but the uploading process usually takes about one minute–depending on your file size and bandwidth. You can register for your own CCP account in seconds.
What is the pre-submission process?
Preparing and uploading your FDA pre-submission meeting request is only the first step of the process. You will receive an automated email confirming that your pre-submission was successfully uploaded. Then, you will receive an automated letter via email that gives you the assigned Q-sub number. You will also receive an automated email notifying you that the pre-submission was accepted, or the FDA reviewer will contact you if changes are needed. The FDA reviewer assigned will usually contact you by email within the first three weeks to schedule a teleconference if you request one. Still, the date/time offered usually does not match the availability of the FDA team, and alternate dates/times may be offered.
You will receive an email response from the FDA for each of your questions within 70 days of receipt by the FDA. If you requested a teleconference, it would typically be about 75 after receipt of the FDA pre-submission meeting request. Your team needs to prepare a detailed discussion plan for the one-hour teleconference. A slide deck is highly recommended to facilitate communication but is not required. If you provide a slide deck, you should email it to the reviewer before the meeting. You must also provide a copy of the slide deck with your meeting minutes. At the beginning of the teleconference, someone from your team must commit to submitting draft meeting minutes to the FDA within 15 days. The FDA will reply with acceptance of your meeting minutes, or they will provide an edited version. It is also common to submit a supplement FDA pre-submission with detailed protocols and new questions for the FDA.

Pre-subs are a valuable communcation tool between Device Mfg and FDA. It can take the guess work (the will or won’t the FDA questions) out of each device/project.
Rob: It would be awesome to see an example of the new PreSTAR.
Hi Nellie,
Thank you for the comment. My first pre-sub using the PreSTAR was just uploaded for a client I referred to you…after an NDA, maybe they will give you a look. In the 4-part webinar series we are actually filling in a hypothetical device PreSTAR to provide people with an example. We will be asking people to vote to select a product this weekend.
Rob
Pingback: FDA Pre-Submission Format And Content Requirements - Plato Data Intelligence
I appreciate your guidance consistently. I have a question regarding my upcoming RTA and AI submissions scheduled after October. I would like to inquire whether these submissions, which were originally planned as eCopy, need to be converted to eSTAR in accordance with the new regulations.
I would greatly appreciate it if you could provide me with feedback on this matter.
Thank you and have a great day!
Great question Heather. No, you do not need to update your submissions to the eSTAR template if you already submitted the 510(k) or De Novo and it is in the review process or currently on hold. Only new submissions submissions that are submitted to the FDA after October 1, 2023 are required to be submitted with the eSTAR template. If fact, even after October 1, 2023 you can still use the eCopy process for responding to the FDA with RTA responses and AI responses. You might consider reviewing the eSTAR template for the section(s) that you are struggling with in the RTA response. The eSTAR might be helpful in preparing your response.