In this article, you will learn how we created a device description template that can be used for US FDA and CE Marking submissions.

Webinar Training
Medical Device Academy also created a webinar on completing this device description template.

This device description template addresses the FDA Refusal to Accept (RTA) guidance document requirements. The template also serves as a summary technical document (STED) for submission to a Notified Body for CE Marking. You would think that it’s tough to screw up the device description, but the FDA screening reviewer is completing a new refusal to accept (RTA) checklist. That checklist has specific requirements for a device description. If you copy the device description from your draft IFU, you will probably receive an RTA letter on Day 15 of the RTA screening process. The review “clock” is reset to zero, and you have to revise your device description and re-submit.
Note regarding changes in the device description template:
The RTA Checklist is no longer relevant for 510k submissions. 510k must now be submitted using the new FDA eSTAR submission templates. This template is your attachment to meet the “Comprehensive Product Description and Principles of Operation Documentation” requirement. However, the FDA now conducts a technical screening rather than completing the RTA checklist. The section numbers are also no longer applicable in a 510k submission.
There are four specific requirements (questions 9-12) in section “B” of the RTA checklist, which is titled “Device Description.” In addition, there are similar requirements for inclusion in a device description for technical files and design dossier submissions to Notified Bodies. Rather than creating two different device description documents, I prefer a template that addresses each regulatory requirement in a single controlled document. Therefore, I created a template for the 510k submission device description with the following headings for Section 11 of the 510k submission:
- Product or Trade Name
- General Description – The general description must be consistent with the device description in the labeling, and this section of the document is intended to address section 13 of the refusal to accept (RTA) checklist.
- Indications for Use – We recommend keeping this separate section of the device description. You should copy the content of FDA Form 3881 verbatim, or the reviewer will indicate that your submission is inconsistent. In the eSTAR, the indications for use can be automatically populated in the 510(k) Summary from FDA Form 3881. We also have a webinar on indications for use.
- List of Devices – A list and description of each device for which a 510(k) clearance is requested in the submission. The list may refer to models, part numbers, sizes, etc. This document section addresses section 14c of the refusal to accept (RTA) checklist. Combining this section with section 3 of the template may be helpful, providing a table with a UDI device identifier for each product listed (if available).
- Intended Patient Population – The medical condition to be diagnosed and/or treated, and other considerations such as patient selection criteria.
- Intended User(s) -Each potential user group should be identified, and it should be stated if the device is intended for use by a healthcare professional, a layperson or both. Finally, this section should indicate if the device is for prescription use, over-the-counter use or both.
- Principles of operation of the device – This document section addresses section 14a of the refusal to accept (RTA) checklist.
- Risk class and applicable classification rule – This is only required for CE Marking, and we typically exclude this from our device description in a PreSTAR or eSTAR submission for the FDA. If you are preparing a device description for CE Marking, the risk classification is based on Annex VIII (MDR) and MDCG 2021-24.
- Conditions of Use (i.e., Environment of Use) – A description of proposed conditions of use, such as surgical technique for implants; anatomical location of use; user interface; how the device interacts with other devices; and/or how the device interacts with the patient. This section should also state where the product is used (i.e. home use or clinical use). This section of the document is intended to address section 14b of the refusal to accept (RTA) checklist.
- Novel Features – This is required for CE Marking, but in a 510k submission, we are trying to demonstrate how the subject device is equivalent to the predicate instead of highlighting novel features. Therefore, any novel features in a 510k should be technological characteristics that you can provide a justification and/or testing to support. This section is not for marketing.
- Components – Description of components, accessories, other medical devices, and other products that are not medical devices intended to be used in combination with the device. The 510k number should identify each component/accessory part of a previous submission. Any component(s)/accessory(s) that have not received prior clearance should also be identified. Sometimes, a side-by-side table for USA and EU markets is needed for accessories that are used in different markets. This document section addresses section 12a, b, and c of the refusal to accept (RTA) checklist.
- Accessories – Description of accessories, other medical devices, and other products that are not medical devices, which are intended to be used in combination with the device. Each accessory that was part of a previous submission should be identified by the 510k number. Any accessory(s) that have not received prior clearance should also be identified. This document section addresses sections 15a, b, and c of the refusal to accept (RTA) checklist. In the eSTAR, there is a subsection at the end of the Product Description section titled “System/Kit Components and Accessories.” If your device is intended to be marketed with multiple system/kit components or accessories, then you must attach a list of those components or accessories in the PreSTAR or eSTAR (see screenshot below).

- Configurations/Variants – Description or a complete list of the various configurations/variants of the device that will be available
- Functional Elements – General description of the key functional elements, formulation, composition, and functionality—including labeled pictorial representations (e.g., diagrams, photographs, and drawings)
- Raw Materials – This section is a duplicate of the section included for biocompatibility. Any raw materials incorporated into components of the device that are intended to make direct contact with the human body or indirect contact with the body should be listed. Any colorants should be included in this list of raw materials.
- Technical Specifications – Technical specifications of the device and any variants and accessories that would typically appear in the product specification are made available to the user, e.g., in brochures, catalogs, and the like.
- Drawings, Schematics, Illustrations, Photos and/or Figures – Representative engineering drawing(s), schematics, illustrations, photos, and/or figures of the device. This document section addresses section 14d of the refusal to accept (RTA) checklist. These drawings, photos, videos, etc. can be attached to the PreSTAR or eSTAR using the button shown in the screen capture below. The FDA requests schematics, drawings, or photos of the product packaging as an attachment to the PreSTAR or eSTAR as well. The best thing to attach for this requirement is a work instruction that illustrates where labeling is applied to the device and the different levels of packaging. This will describe the packaging and how the device is packaged and labeled.

- Similar & Previous Generations of the Device – Reference to similar and previous device generations. Ensuring these devices are included in the clinical evaluation report is important. If submitting a 510k submission, you want to ensure that any devices are registered and listed with the US FDA in the same product category. Creating a table that organizes the “similar” devices by intended use and technological characteristics may be necessary for a device with multiple predicates.
- Requirements Specific to the Special Controls Guidance Document – This template section addresses section 12 of the refusal to accept (RTA) checklist. When you complete the classification section of the PreSTAR or the eSTAR, the PDF template should automatically identify and Special Controls Guidance Documents that are applicable (see example below).
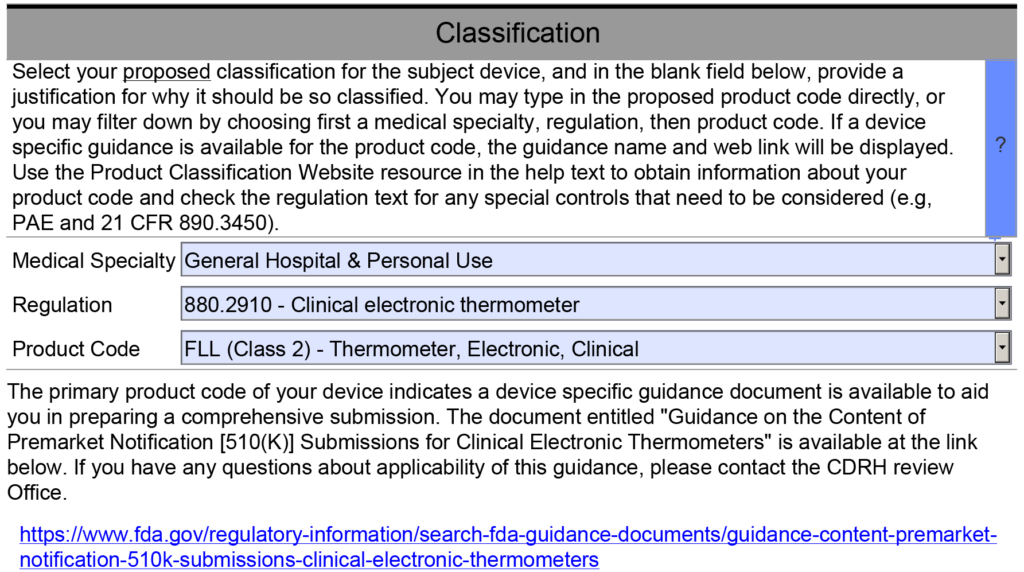
The last section of the device description is for any unique requirements specific to the special controls guidance document for the product classification I am working on. However, most of the requirements for a device description are met by the previous items in my outline. Therefore, I created a table that outlines each requirement of the Special Controls Guidance Document, and I provided a cross-reference to the section of the outline that includes this requirement. If requirements are not covered elsewhere in the document, I address them in the table. If there is no Special Controls Guidance Document, then I state that no Special Controls Guidance Document exists for the product.
Thanks for the information.
It is useful and very appropriate to us (as we are considering adding incremental functionalities to our medical instrument).
Please keep these articles coming.
Your guidance is valued and appreciated.
Thanks.
J.D.
Jean-Denis Crepeau
QA/RA Manager
Annidis Corporation
100 Maple Grove
Ottawa, Ontario, Canada
K2V 1B8
(613) 595-1800 xt 2017.
Thank you for the positive feedback Jean-Denis. I also have a suggestion box (http://robertpackard.wpengine.com/suggestion-box/) where you can enter suggestions for future blogs, webinars and workshops.
Pingback: 4 keys for preparing a medical device submission to the FDA