This webinar explains how to create a design history file (DHF) complaint with 21 CFR 820.30j and ISO 13485:2016, Clause 7.3.10.
This presentation includes specific documentation updates to comply with ISO 13485:2016 that was released on March 1, 2016 and how to integrate your design process with preparation of regulatory submissions such as a 510k or CE Marking submission.
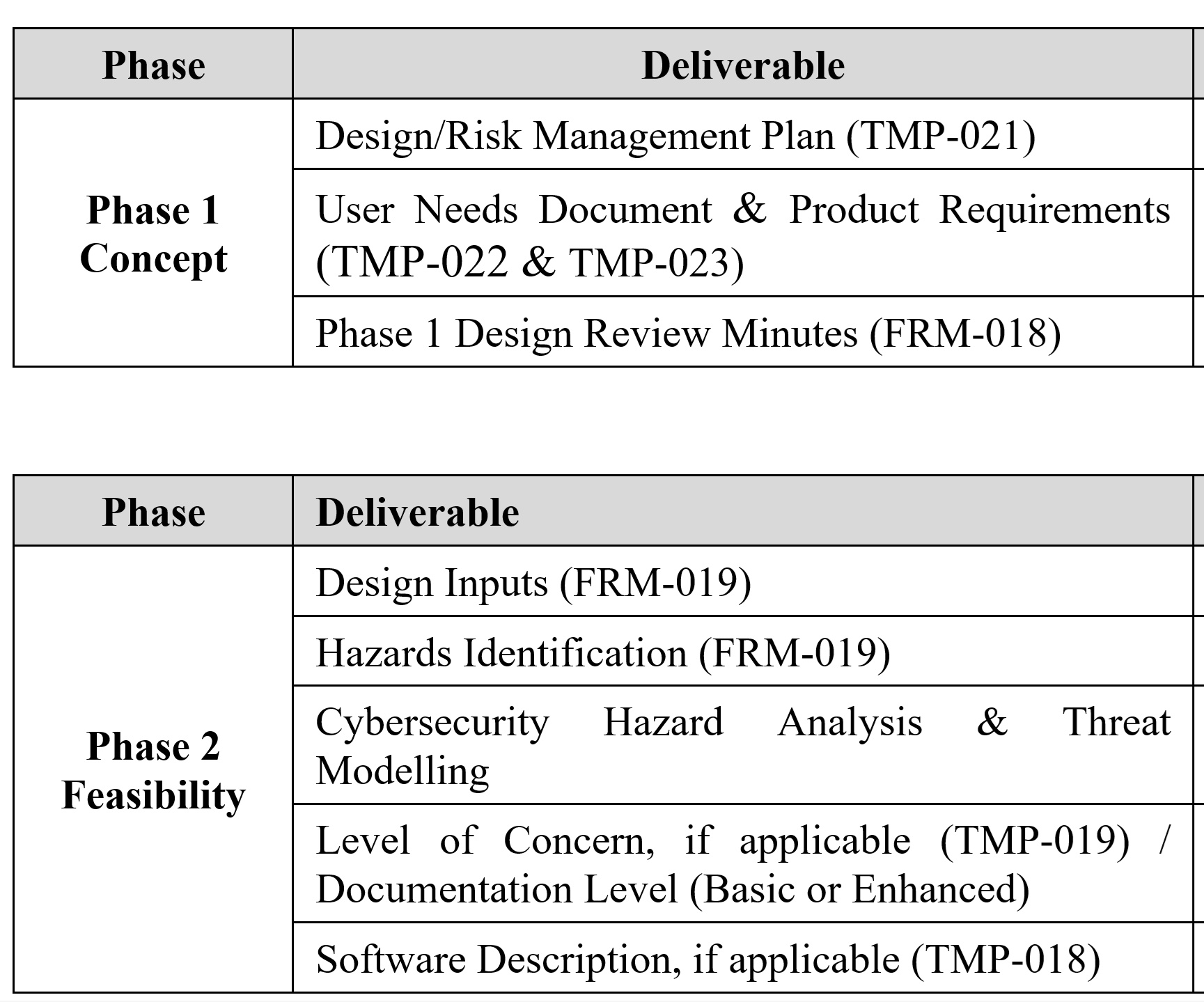
Design History File (DHF) for 21 CFR 820.30j compliance
The most frequent category of FDA Form 483 inspection observation is design controls (i.e., 21 CFR 820.30). There are 10 different sections of the design controls requirement. Manufacturers that do not have design controls in place will frequently receive multiple observation findings during the same inspection–all related to design controls. The most common design control observations are:
- A procedure for design and development has not been established in accordance with 21 CFR 820.30a
- A procedure for design transfer has not been established in accordance with 21 CFR 820.30h
- A procedure for design changes has not been established in accordance with 21 CFR 820.30i
- A design history file (DHF) has not been established in accordance with 21 CFR 820.30j

If a manufacturer has no procedure for design controls, then the manufacturer could receive 4 different observations on FDA Form 483.
ISO 13485:2016 Requirements for Medical Device Files your Design History File (DHF)
On March 1, 2016, the 2016 version of ISO 13485 was released. The new version of the Standard now requires procedures for design transfer, design changes, and design and development files to be further harmonized with US regulatory requirements. Then, on February 2, 2024, the FDA released the QMSR–which goes into effect on February 2, 2026. The new QMSR does not include the term Design History File (DHF). Instead, the FDA is incorporating the requirements of ISO 13485 by reference. Specifically, that includes Clause 4.2.3 for Medical Device File. Therefore, this presentation was created to specifically identify changes needed to your design controls procedure in order to comply with ISO 13485:2016 and the QMSR.
When is the Design History File (DHF) Webinar?
This webinar will be hosted live on September 26, 2024 @ 10:30 a.m. ET. If you purchased the webinar before September 26, you will receive login information to participate in the live webinar. The webinar will be hosted on Streamyard.com. If you cannot participate in the live webinar, please send us your questions in advance so that we can be sure to address your questions in the live webinar. You will be able to download the recording from the Dropbox folder after the live webinar and you can watch it as many times as needed. You will receive:
- a native slide deck for the webinar (30+ slides)
- our combined design and risk management plan template (TMP-021)
- a link to download a recording of the webinar (60+ minutes)
All content deliveries will be sent via AWeber emails to confirmed subscribers.
Do you have questions about design history files?
If you have any generic questions about creating a design history file or the new ISO 13485:2016 requirements for a design and development file, please email me at rob@fdaestar.com. I will use your questions as material for webinars and future blogs. During live webinars, you are only able to submit questions via the chat box. If you have company-specific questions, please send me a request to set up a private call to discuss your specific issues.
Design History File (DHF) Webinar available for $129.00:
Exam and Training Certificate available for $49.00:

VIEW OUR PROCEDURES – CLICK HERE OR IMAGE BELOW:

About Your Instructor

Rob Packard is a regulatory consultant with ~25 years of experience in the medical device, pharmaceutical, and biotechnology industries. He is a graduate of UConn in Chemical Engineering. Rob was a senior manager at several medical device companies—including the President/CEO of a laparoscopic imaging company. His Quality Management System expertise covers all aspects of developing, training, implementing, and maintaining ISO 13485 and ISO 14971 certifications. From 2009 to 2012, he was a lead auditor and instructor for one of the largest Notified Bodies. Rob’s specialty is regulatory submissions for high-risk medical devices, such as implants and drug/device combination products for CE marking applications, Canadian medical device applications, and 510k submissions. The most favorite part of his job is training others. He can be reached via phone at +1.802.281.4381 or by email. You can also follow him on YouTube, LinkedIn, or Twitter.